Hi all:
The tension behavior of the CeO2 is simulated through lammps. The input file is next:
units metal
atom_style charge
boundary p p p
read_data gdc5.data
neighbor 2.0 bin
neigh_modify delay 0 every 20 check no
kspace_style ewald 1e-6
pair_style buck/coul/long 8.0
pair_coeff 1 1 22764.3 0.1490 20.37
pair_coeff 1 2 1017.40 0.3949 0.000
pair_coeff 1 3 1336.80 0.3551 0.000
pair_coeff 2 2 0.00000 1.0000 0.000
pair_coeff 2 3 0.00000 1.0000 0.000
pair_coeff 3 3 0.00000 1.0000 0.000
velocity all create 20 87287 dist gaussian
fix 1 all npt 20 20 0.1 aniso 0.0 0.0 0.0 0.0 0.0 0.0 0.1 drag 1.0
thermo 100
timestep 0.001
run 10000
unfix 1
fix 1 all npt 20 20 0.1 aniso 0.0 0.0 0.0 0.0 NULL NULL 0.1 drag 1.0
variable i loop 80
label loop
print step=$i
displace_box all z delta -0.1 0.1 units box
run 1000
next i
jump in.tension loop
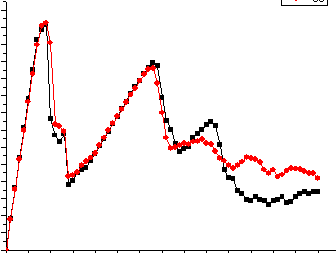
The PDB is used for the system. But the stress-strain curve shows that the boundary conditon is free.(The curve is in the annex). Who can give me some suggestion? In my opinion, the infile is correct, but I cann’t find the reason! Thanks for help! Thanks!
|
1.bmp (249 KB)
{kind=link}