Dear LAMMPS community,
I am interested in simulating ionic transport of magnesium electrolytes in poly(ethylene oxide) (PEO) in LAMMPS. Borodin and Smith demonstrated that using a many body polarizable force field is necessary for accurate dynamics, and developed a polarizable force field for PEO [1]. They implemented their force field using Lucretius MD code, which no longer exists (at least for free). Is the best/only way to implement this in LAMMPS to write a pair_style? Due to the form of the potential it would just consist of adding the polarization term to the buck/coul force field that already exists, I think.
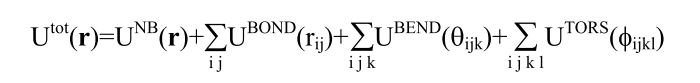
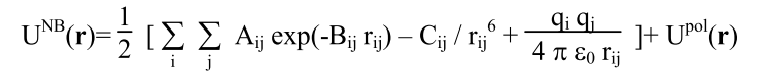
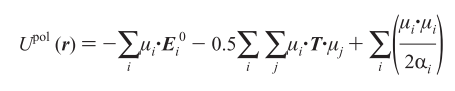
Or would a different preexisting potential that includes polarization effects be better suited? Neither reaxFF or COMB3 support all the atoms types needed so some force development seems inevitable. Any suggestions are welcomed.
[1] Borodin, Oleg, and Grant D. Smith. "Development of quantum chemistry-based force fields for poly (ethylene oxide) with many-body polarization interactions."The Journal of Physical Chemistry B 107.28 (2003): 6801-6812.
Best regards,
Brandon Wood
Dear LAMMPS community,
I am interested in simulating ionic transport of magnesium electrolytes in
poly(ethylene oxide) (PEO) in LAMMPS. Borodin and Smith demonstrated that
using a many body polarizable force field is necessary for accurate
dynamics, and developed a polarizable force field for PEO [1]. They
implemented their force field using Lucretius MD code, which no longer
exists (at least for free). Is the best/only way to implement this in
LAMMPS to write a pair_style? Due to the form of the potential it would
just consist of adding the polarization term to the buck/coul force field
that already exists, I think.
adding such a dipole polarization term could indeed be implemented as a
pair style and then combined with the buck/coul potentials via pair_style
hybrid/overlay
the important question is the range of the polarization interaction, i.e.
whether it needs to include long-range electrostatics or not. if they do,
things get complicated and probably an approach using a fix doing the
self-consistent part of the calculation in addition would be required
(similar to how it is done in COMB3).
Or would a different preexisting potential that includes polarization
effects be better suited? Neither reaxFF or COMB3 support all the atoms
types needed so some force development seems inevitable. Any suggestions
are welcomed.
to the best of my knowledge, the only similar feature in LAMMPS is support
for charge equilibration, which is applied to point charges and uses the
wolf-summation approach to approximate long-range electrostatics. i do
know of several attempts to implement the point dipole polarizable model of
Paul Tangney and Sandro Scandolo into LAMMPS, but so far have not seen any
usable code.
axel.