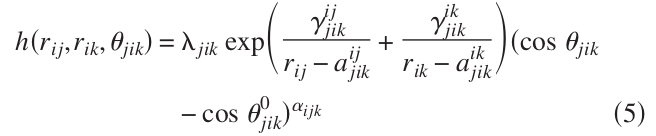I’m trying to apply a set of sw potential parameters in Lammps for a Si-O-C-F system. See below for the original journal article:
Smirnov, V. V., et al. “Molecular-dynamics model of energetic fluorocarbon-ion bombardment on SiO 2 I. Basic model and CF 2±ion etch characterization.” Journal of applied physics 97.9 (2005): 093302.
The form of the 3-body interaction of SW potential they used in the paper is slightly different than what we have in Lammps (see below)
All the other variables can be converted to Lammps style, however, the power of the (cos\theta - cos\theta^0) term is a variable alpha_ijk, while in Lammps it is a constant 2.
Is there any way I can modify the sw potential so I can customize that power variable?
It seems like in the source code (lammps/src/MANYBODY/pair_sw.cpp), at line 504, we can modify that line in this way for example:
In LAMMPS variants of a potential are usually introduced by creating a derived class and providing only additional or modified class member variables and functions in the derived class. See for example the “sw” and “sw/mod” pair styles and their corresponding sources in src/MANYBODY/.
In this particular case you would extend the PairSW::Param struct to contain the additional per atom type triple parameter, and also declare the “read_file()” function in PairSW as “virtual”.
The new derived class would then only need to provide overrides for the “read_file()” and “threebody()” functions that parse and use the added parameter.
I was able to do some research on the source code based on your suggestions. It seems like NPARAMS_PER_LINE controls the number of parameters to be used in this class, and all the parameters (for the base class and all the derived class) are first introduced in the base class.
For example, the Param struct in pair_tersoff.h has 33 members while it’s using 17 of them, and pair_tersoff_zbl.h would use 4 extra members.
That means if I want to add one parameter to the derived class of pair_sw.h, I need to add it in the base class first. Is there any way I can extend Param struct in the derived class only so that I don’t need to modify the base class? The reason is that if I update LAMMPS using git pull, I may get conflict if I modify the base class.
This is not a problem since, a) those constants are static class members, so you can define them to a different value in your derived class, b) you need to write a modified version of the parser to accept this additional parameter anyway, c) for as long as only members are added to the Params struct there is no conflict.
Theorectically, yes, but it would be very unpractical since it would stop you from sharing code.
This is very unlikely and in any case, those conflicts would be easily resolved. There are many ways that changes to the PairSW class or any of the LAMMPS core code can break your custom pair style without(!) merge conflicts from upstream. The best way to protect yourself from such unexpected and undesired breakage is to create a suitable unit test yaml file: 4.10. Adding tests for unit testing — LAMMPS documentation
And enabling and running tests with ctest.
This is also the reason why it is beneficial to have extended/modified/custom functionality submitted for inclusion into the LAMMPS distribution, since we do use automated testing on any submitted code, so it will be difficult to make changes that will create merge conflicts, break compilation, or change results of the tests in the unittest tree (provided one or more tests have been submitted with the added code).
I was able to create a derived class (called sw/mod/a and PairSWMODA) based on your suggestion. One extra parameter “poweralpha” was added to the Param struct in pair_sw.h and methods read_file() and threebody() were declared as virtual, which were then overridden in the derived class. LAMMPS was re-complied successfully.
I also did a simple test to make sure the sw/mod/a potential works as expected. I used the benchmark file (LAMMPS/bench/POTENTIALS/in.sw) for silicon crystal. File in.sw was copied and the potential was changed to sw/mod/a. The potential file Si.sw was also copied with one extra column added for alpha (value=2.0).
The results of the test case match the benchmark very well. However, the test case is slower than the benchmark, which is probably due to std::pow(var, 2.0) is less efficient than var*var in C++.
Everything looks good now. Thanks again for your help!
If poweralpha will always be an integer, then there is a MathSpecial::powint() function defined in the “math_special.h” header that is significantly faster than std::pow().
From the paper I’m referencing, poweralpha will only be an integer 2 for some Si-X-Y (Si as the center atom) pairs. For other pairs, it could be some value like 2.708 for C-F-O.
I am also trying to extract the potential function from this file, but I am a beginner and I cannot understand these data. So I want to ask if you can help me. If you can share the Si-O-C-F potential function file with me, I will thank you very much