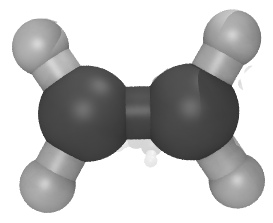
Moltemplate version 1.20 has been released.
I'm announcing it here because a major feature has been added by Jason
Lambert. You can now build molecules using moltemplate with automatic
OPLSAA force-field assignment. (Atom charge is also assigned
automatically by atom type by default.)
Jason's contribution means that moltemplate is now probably one of the
easier (non-graphical) ways to prepare OPLSAA simulations for LAMMPS.
You can go to moltemplate.org and download the examples. There is an
example how to build a simple molecular system located in this
directory:
all_atom_examples/OPLSAA_force_field_examples/ethylene
Here is a brief outline of this (relatively simple) process.
1) Create a file which defines a molecule in moltemplate (.lt) format.
The atoms in that file should use OPLSAA atom name conventions. (See
"ethylene.lt" file attached for a simple example.)
2) Edit "oplsaa.txt" file (located in the "common/oplsaa" directory)
to delete atoms (lines beginning with the word "atoms") which you
don't need in your simulations. Then save the new file as
"oplsaa_subset.txt"
3) Run Jason Lambert's "oplsaa_moltemplate.py" script:
./oplsaa_moltemplate.py oplsaa_subset.txt
4) Run moltemplate:
moltemplate.sh system.lt
This will create 5 files
system.data (coordinates and topology)
system.in.init (atom_style, force-field styles)
system.in.settings (force-field parameters, groups, constraints etc..)
system.in.charges (atomic charges)
system.in
You will have to edit the "system.in" file to specify the timestep,
and run-time fixes (nvt, npt) that you wish to use during your
simulation. Otherwise these files are ready to run in LAMMPS.
Note: The OPLSAA implementation in moltemplate currently uses
pair_style lj/cut/coul/long 10.0 10.0
If this is not appropriate for OPLSAA, please let me know...
Disclaimer: Just because the program is easy to use, it does not mean
that it actually works. This code, and this example were created
recently. Apart from running a short simulation (which did not
crash), we have not tested it rigorously for accuracy.
As always, please let us know if there are bugs or problems.
I hope this is useful.
Thanks Jason!
Andrew
P.S. AMBER-GAFF support was added in 2013-8, however it does not
assign atom charge (and I confess it is still a little bit awkward to
use).
ethylene.lt (1.31 KB)
system.lt (511 Bytes)