Dear all,
I want to calculate self diffusion coefficient for water model(SPC) it has 300 water molecule.
I know that I can calculate the diffusion coefficient by using Einstein relationship with MSD.
Experiment result is 2.3 E-09 m2/s (roughly), but my result is about 1/100 of the expected result.
I ran this model 100ps in npt to get equilibrium(save as write_restart) and ran 200ps more in nvt with read_restart command.
Units
units real
Resion
boundary p p p
Force type
atom_style full
pair_style lj/cut/coul/cut 9 9
bond_style harmonic
angle_style harmonic
Atom definition
read_data data_300W.txt
velocity all create 300 8343 dist gaussian units box
Neighbor list
neighbor 2.0 bin
neigh_modify every 10 delay 0 check yes
Calculation
thermo_style custom step pe ke etotal temp press vol density
thermo 10
Minimization
minimize 1.0e-5 1.0e-7 1000 100000
Timestep
timestep 1
Fix molecular
fix 1 all shake 1.0e-4 10 0 b 1 a 1
Dynamic type
fix 2 all npt temp 298.15 298.15 100 iso 1 1 100
Start running
run 100000
Save the restart file
write_restart restart300W.txt
Units
units real
Resion
boundary p p p
Force type
atom_style full
pair_style lj/cut/coul/cut 9 9
bond_style harmonic
angle_style harmonic
Atom definition
read_restart restart300W.txt
Neighbor list
neighbor 2.0 bin
neigh_modify every 10 delay 0 check yes
Calculation
thermo_style custom step pe ke etotal temp press vol density
thermo 10
Timestep
timestep 1
Fix molecular
fix 1 all shake 1.0e-4 10 0 b 1 a 1
Dynamic type
fix 2 all deform 1 x final 0.0 20.992339 y final 0.0 20.992339 z final 0.0 20.992339
#fix 3 all npt temp 298.15 298.15 100 iso 1 1 100
fix 3 all nvt temp 298.15 298.15 100
#fix 3 all nve
Group
group ox type 1
Compute
compute 1 ox msd
#compute 2 all rdf 1000 1 1 # O-O
#compute 3 all rdf 1000 2 2 # O-H
#compute 4 all rdf 1000 1 2 # H-H
Variable
variable msd equal c_1[4]
variable fs equal 1*step
variable Temp equal “temp”
variable Vol equal “vol”
variable density equal “density”
Print results and make files
fix 7 all print 10 “{fs} {msd}” file water.msd screen no
Run a simulation
run 200000
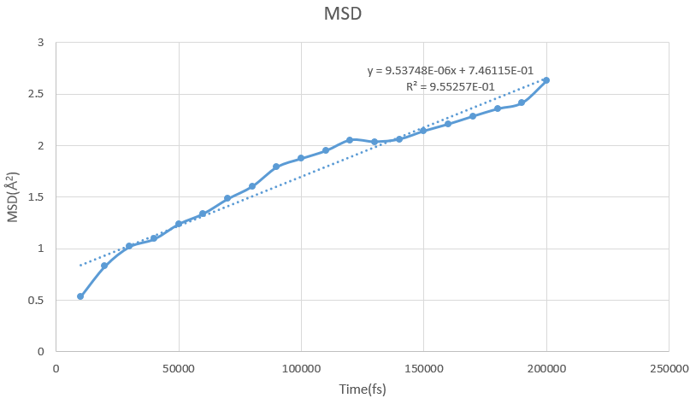
Above are input file and result, MSD is too small than I expect. Can I have any suggestion ?
Thanks,