Hello all,
I did molecular dynamic simulations of the crosslinking process of epoxy in Materials Studio. To continue the simulation in LAMMPS, I created the data file by saving the car/mdf file of the results and converted using “msi2lmp” function with PCFF. For my initial simulations, I used LAMMPS 11aug2017-MPI version. I faced no error in reading the data file (except a warning of an inconsistent image flag because of the periodic graphene in the system).
Hello all,
I did molecular dynamic simulations of the crosslinking process of epoxy in Materials Studio. To continue the simulation in LAMMPS, I created the data file by saving the car/mdf file of the results and converted using “msi2lmp” function with PCFF. For my initial simulations, I used LAMMPS 11aug2017-MPI version. I faced no error in reading the data file (except a warning of an inconsistent image flag because of the periodic graphene in the system).
But now I have realized that in the newer version of LAMMPS, I have the following error:
“Invalid atom ID in Dihedrals section of data file”
I searched the mail list for the reasons! I found Dr. Axel Kohlmeyer response:
the old version doesn't complain, because it does not have the check
for invalid topology data.
i don't know why msi2lmp produces these files, but i suspect that
already the topology data you have as input may be incorrect.
Now here are my questions:
1- What is the origin of the problem? Does the problem come from the msi2lmp function or the way that for example Materials Studio export car/mdf file?
difficult to say in this generality.
2- Does anyone know any other way to solve this problem? Because it is really hard to create the data file manually for a class2 forcefield.
i would look at the exact data that is causing the error and try to
understand what is wrong here. it may depend on the force field
specifics.
3- If we use older versions of LAMMPS, and find no error for the simulations, then should we concern about the results?
yes. avoiding an issue without understanding it list like holding a
grenade with a pulled pin in our hand. nothing will happen initially,
but as soon as you drop it, you are in big trouble.
axel.
Dear Axel,
Thank you so much for your quick response.
Do you think attaching the Data file can be helpful for finding the problem? This is the complete error message:
Dear Axel,
Thank you so much for your quick response.
Do you think attaching the Data file can be helpful for finding the problem? This is the complete error message:
the error message indicates, that you have dihedral definitions where the same atom appears more than once. you can write a simple script to detect the offending lines. here is one that i wrote for such a purpose:
#/usr/bin/perl -w
my $in_dihed=0;
my $line = 0;
my @words = ();
while (<>) {
++line;
chomp(_);
if (/\sDihedrals\s(|#.*)$/) {
print(“Got dihedrals section. Line $line\n”);
in_dihed = 1;
}
if (/\s*(Bonds|Angles|Impropers)\s*(|\#.*)/) { $in_dihed = 0; }
if (in_dihed) {
@words = split /[ \t]+/, _;
if ($#words eq “6”) {
print (“Atom-ID 1 == 2. Line $line\n”) if ($words[3] eq $words[4]);
print (“Atom-ID 1 == 3. Line $line\n”) if ($words[3] eq $words[5]);
print (“Atom-ID 1 == 4. Line $line\n”) if ($words[3] eq $words[6]);
print (“Atom-ID 2 == 3. Line $line\n”) if ($words[4] eq $words[5]);
print (“Atom-ID 2 == 4. Line $line\n”) if ($words[4] eq $words[6]);
print (“Atom-ID 3 == 4. Line $line\n”) if ($words[5] eq $words[6]);
}
}
}
Thank you so much for the help.
I will work on your comments. Hope to find the problem.
Bests,
Abolfazl
Dear Axel,
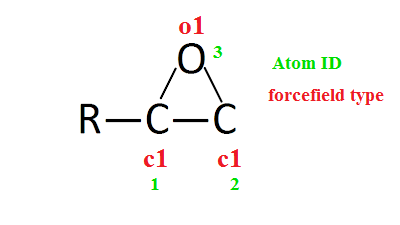
I think I found the problem! My initial structure has an epoxide ring (a ring containing three atoms). Using msi2lmp.exe function defines dihedrals (AngleAngleTorsion, etc.) coefficients in the data file with the same first and forth atom ID! To be more clear, see the enclosed image:
For example in dihedral coefficient section, there are a dihedral coeffs for:
c1 c1 o1 c1
and based on atom IDs in dihedrals we have,
Num Type 1 2 3 1
So, we have the same atom IDs in a line!
Now, is that a bug with msi2lmp.exe function?
Bests,
Abolfazl
The msi2lmp program is known to contain bugs. But it was written by
people who long ago stopped maintaining the code. (It also does not
handle "auto-equivalences" present in the CVFF and PCFF force fields.)
Axel has been doing an admirable job to maintain it. But if you are
using COMPASS or PCFF, there are several alternatives:
If you are using the PCFF force field, try using EMC.
If you are using COMPASS, try using either moltemplate or EMC.
Other builders are listed here:
https://lammps.sandia.gov/prepost.html#builders
To my knowledge, none of these programs can build molecules with
unpublished atom types (such as Sp2 carbons) unless you have an ".frc"
file that describes them. (However it does not look like you have
this problem.)
Good luck.
Andrew
Thank you Jewett.
No one can ignore the expertise and responsibility of Dr. Axel Kohlmeyer in this regard.
Thank you so much for your help. I will go through your comments, and I hope everything goes well with creating the data file.
Bests,
Abolfazl
yes. msi2lmp does not (yet) check for this case. fortunately, this is easy to add to the code. like with the following patch:
$ git diff
diff --git a/tools/msi2lmp/src/MakeLists.c b/tools/msi2lmp/src/MakeLists.c
index 19924386d…44b9bdd0a 100644
— a/tools/msi2lmp/src/MakeLists.c
+++ b/tools/msi2lmp/src/MakeLists.c
@@ -363,7 +363,7 @@ int count_dihedrals()
if (i != k) {
for (ll=0; ll < atoms[k].no_connect; ll++) {
l = atoms[k].conn_no[ll];
- if ((l != j) && (i != l)) n++
}
}
}
@@ -391,7 +391,7 @@ void build_dihedrals_list()
if (i != k) {
for (ll=0; ll < atoms[k].no_connect; ll++) {
l = atoms[k].conn_no[ll];
- if ((l != j) && (i != l)) {
dihedrals[n ].type = 0;
dihedrals[n ].members[0] = i;
dihedrals[n ].members[1] = j;
diff --git a/tools/msi2lmp/src/msi2lmp.c b/tools/msi2lmp/src/msi2lmp.c
index b0bc7815b…efc929639 100644
— a/tools/msi2lmp/src/msi2lmp.c
+++ b/tools/msi2lmp/src/msi2lmp.c
@@ -2,6 +2,8 @@
Wow!
Thank you very much!
Sure, I will keep you posted.
Best Regards,
Abolfazl
Dear Axel,
I am glad to inform you that the modification worked! I applied the modification to the msi2lmp file, recompiled it, and create the data file! It seems the modification has solved the topological problem and no dihedral parameters were created for a 3-atom ring!
Thank you very much for your time and consideration.
Bests,
Abolfazl