Dear lamps-users,
I’m trying to calculate the migration energy of interstitial Si atom in 3C-SiC lattice. Here I did the neb calculation and the version of my LAMMPS is 22 Aug, 2018.
My input is similar to the example provided by LAMMPS, as shown below:
units metal
dimension 3
boundary p p p
atom_style full
atom_modify map array sort 0 0.0
variable u uloop 20
pair_style tersoff
pair_coeff * * SiC.tersoff C Si
neighbor 0.3 bin
neigh_modify delay 5
create_atoms 2 single 2.1874 4.3480 4.3480
minimize 1.0e-6 1.0e-4 1000 10000
reset_timestep 0
timestep 0.01
thermo 100
fix 1 all neb 1.0
dump 1 all atom 10 dump.neb.$u
min_style quickmin
neb 0.0 0.1 1000 1000 100 final final.isi
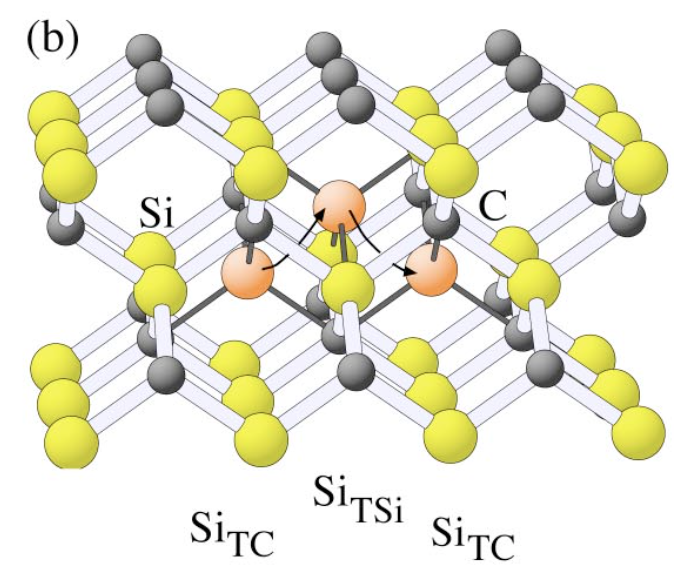
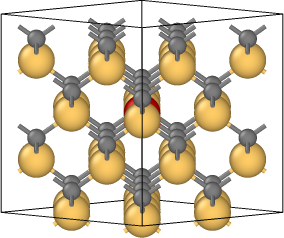
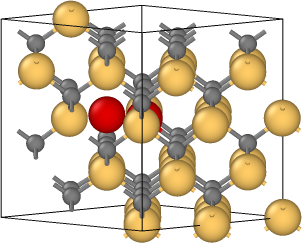
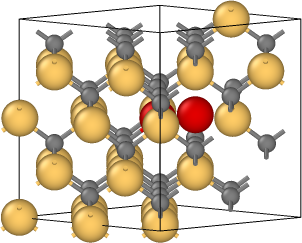
The coordinates in file final.isi are obtained after relaxation. The tetrahedral interstitial atom moves to its first neighbor tetrahedral interstice by a kick-out process that kicks the middle Si to become the defect but itself replaces its position in literature. And I put the initial lattice(relax.data), schematics of the process (kick-out.png), positions of interstitial Si atoms(initial.png, defect_1.png, defect_2.png, and final.isi) in the attachment.
The problem is that my system does not seem to converge, even though I’ve tried to tune the parameters with larger maximum iteration steps, different timesteps, and different numbers of partitions. I also tried to provide the positions of neighboring atoms near the interstitial Si in the final.isi, but I got similar outputs of log files in attachment log.lammps.
I checked the neb questions in the mailing lists but still quite confused. Looking forward to your suggestions.
Thank you.
Best,
Wanzhen
final.isi (48 Bytes)
log.lammps (7.08 KB)
relax.data (11.3 KB)