- The potential in the Kern-Frenkel paper is a hard-sphere square-well with direction-dependent attraction. Here’s a picture from the paper:
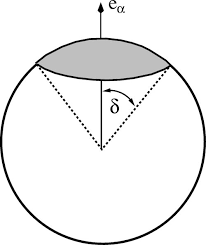
This kind of force-field is more appropriate for a Monte-Carlo simulation, not a molecular dynamics simulation. Molecular dynamics requires you to calculate the force between particles. (Since your particles are not point-like, you also have to worry about torques). But in a square-well potential, the forces (and torques) between particles are either zero or infinity. This means that, if you still plan to use LAMMPS, you would need to integrate over the degrees of freedom of the system using “fix gcmc” (or “fix atom/swap”?) instead of a more traditional method for integrating the equations of motion (such as fix npt, nph, nvt, nve, or langevin).
http://lammps.sandia.gov/workshops/Aug15/PDF/talk_Thompson1.pdf
http://lammps.sandia.gov/doc/fix_atom_swap.html
http://lammps.sandia.gov/doc/fix_gcmc.html
Alternately, you could modify the model so that it uses softer potentials so that the forces are always finite (and hopefully continuous).
Since your particles have multiple attractive patches, you will need to create arrays that store these internal properties of these atoms. You could either try and create your own new atom_style (difficult), or use “fix property/atom” to do this:
http://lammps.sandia.gov/doc/fix_property_atom.html
- The particles in the model are not point-like. Fortunately, LAMMPS can support non-point like particles (using atom_style ellipsoid, for example). LAMMPS uses quaternions to represent the orientational degrees of freedom of these particles.
http://lammps.sandia.gov/doc/atom_style.html
There are examples of pairwise-interactions which depend on the orientation between two particles:
http://lammps.sandia.gov/doc/pair_gayberne.html
If you decide to use LAMMPS to run your simulation, then will have to read how these interactions are implemented (in pair_gayberne.cpp), and create a new pair_style which implements the square-well potential used in the paper.
- The Kern-Frenkel paper also use some form of parallel-tempering (a.k.a. “replica exchange”). In LAMMPS is currently implemented using:
http://lammps.sandia.gov/doc/temper.html
I hope this gets you started.
LAMMPS is not really optimised for this kind of simulation. At the very least, you will have to write your own pair_style (perhaps similar to “pair_gayberne.cpp”.
The only reason that I can think of to try using LAMMPS to run this kind of simulation is that you eventually plan to combine these patchy particles with other more complex molecules or particles and force-fields only supported by LAMMPS. Or if you plan to add molecular details to these patchy particles.
When is LAMMPS useful?
LAMMPS might be more appropriate if you wanted to implement your patchy particles using an atomistic model (perhaps using fix rigid, fix rigid/small, and/or atom_style body.) See pictures below:
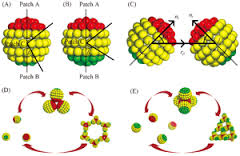
LAMMPS would be especially useful if you plan to add molecular details to these patches, as shown below:

Here’s another example:
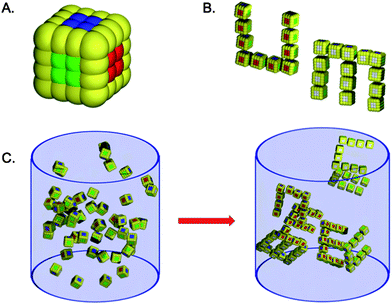
… but all of these models are fundamentally different from the Kern+Frenkel paper (even though they may behave in a physically similar way).
The Kern-Frenkel model a simple elegant model with a small number of parameters. If all you ever plan to do is reproduce the results of the Kern-Frenkel paper exactly, then it is probably more straightforward to simply write your own Monte-Carlo simulation program. (Especially if you only have a small number of particles and don’t need to take advantage of the parallelism that MPI provides. The simulations in the Kern-Frenkel paper only have 512 particles.) But I suspect in the long run, this might not be what you want to do, especially if you plan to add molecular details later.
I hope this helps.
Andrew