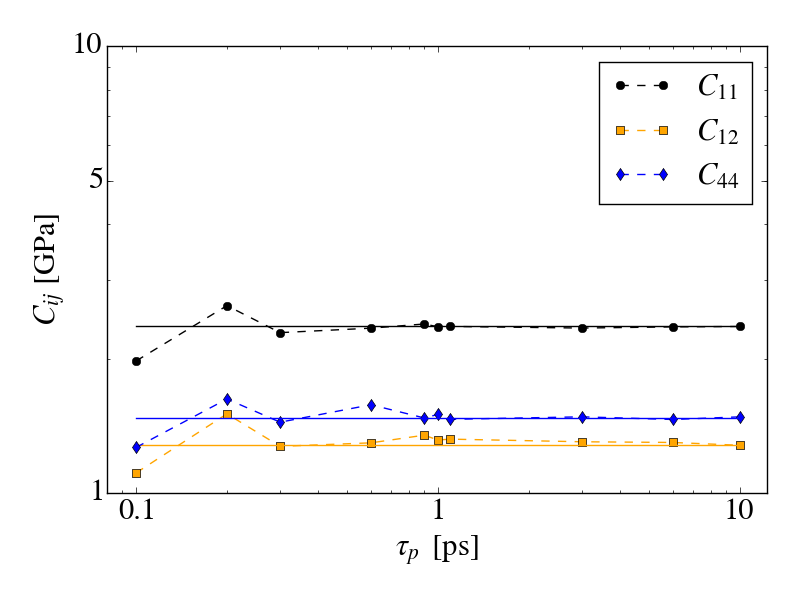
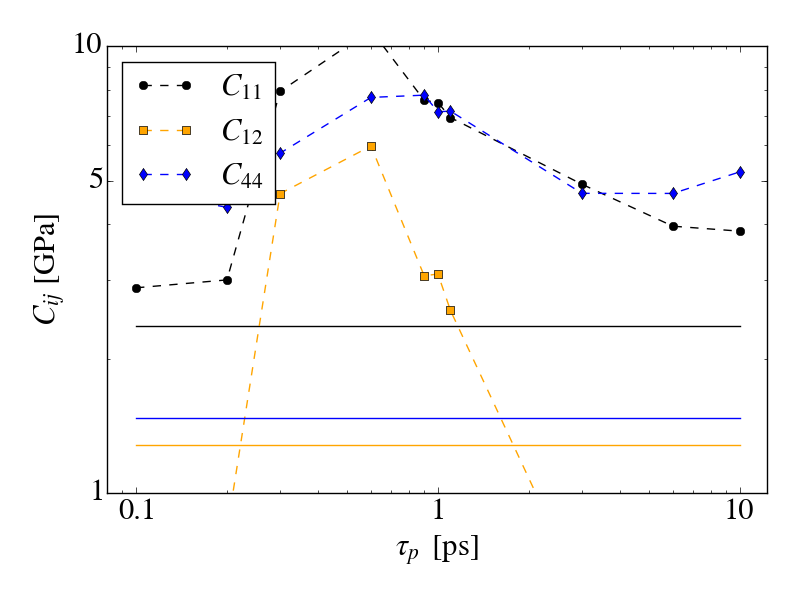
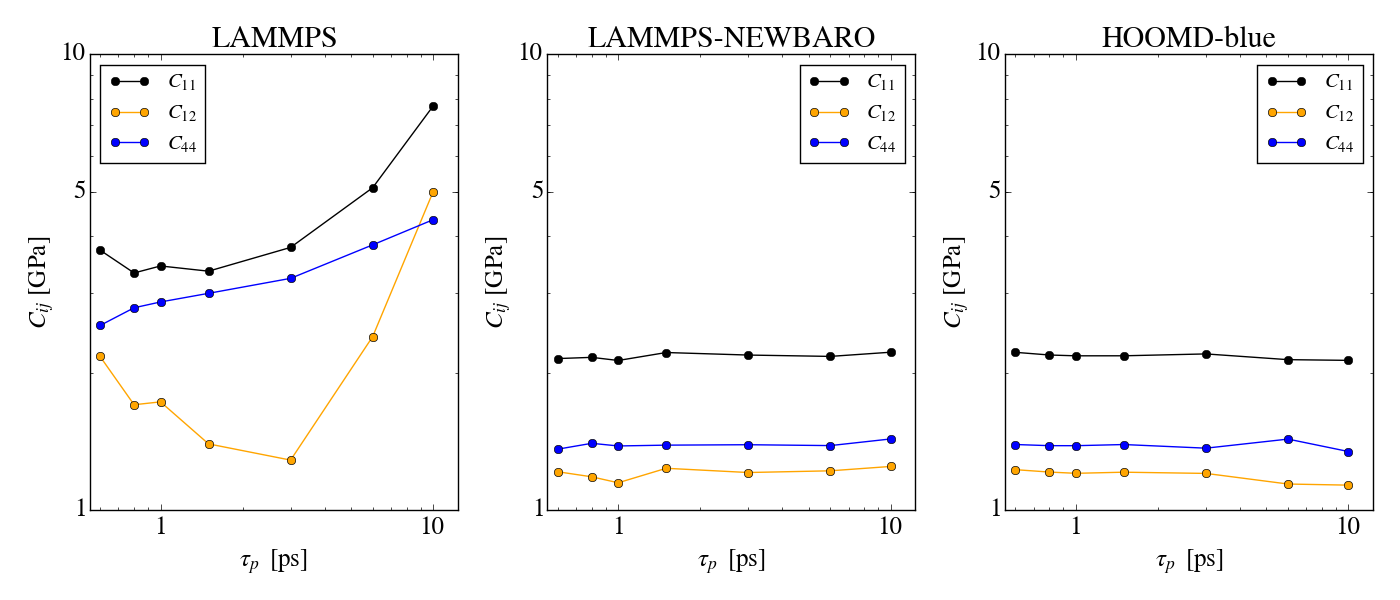
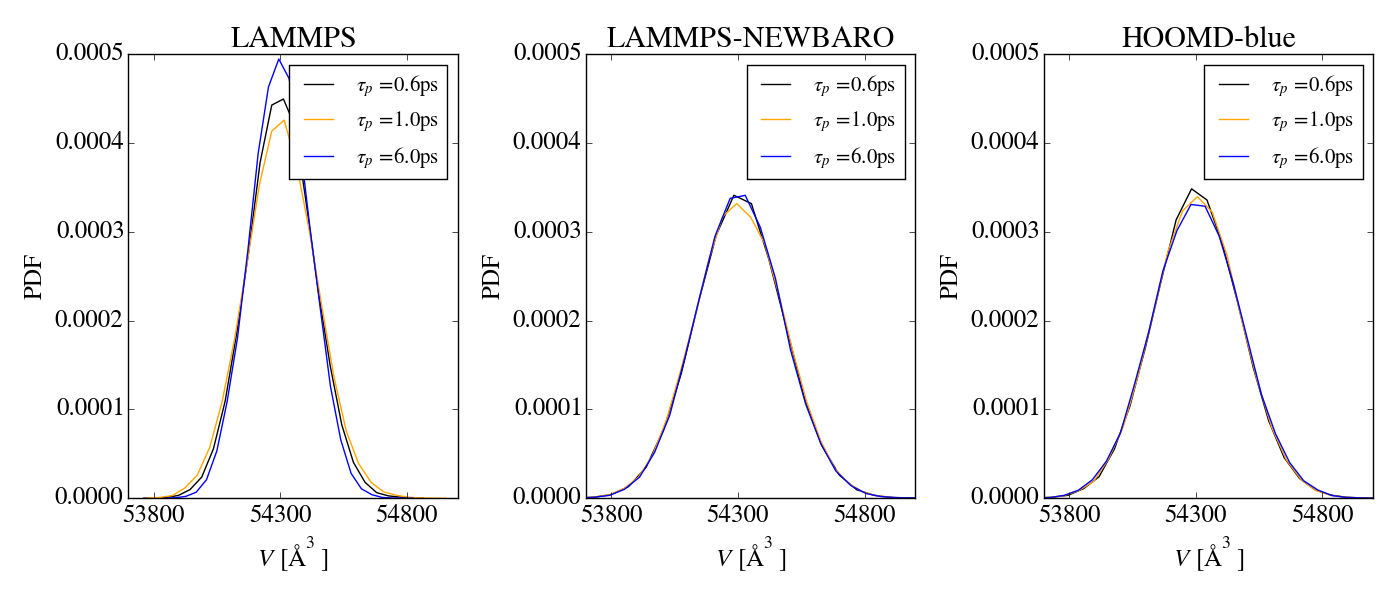
I am running into problems when simulating in the NPT ensemble with
anisotropic cell fluctuations are allowed. In short, when only
considering isotropic cell variations (using the iso keyword), the
conserved quantity is nearly constant and the volume fluctuations seem
correct. As soon as anisotropic cell variations are allowed (using for
example the aniso or tri keywords), there is a clear drift in the
conserved quantity as well as indications that the ensemble is
incorrectly sampled.
The test system is a metal-organic framework, MOF-5 with an initially
cubic unit cell (I am aware that this is not the most simple test
system, any suggestions for a less complicated case are welcome). The
force field used is UFF4MOF (Lennard-Jones pair interactions, no
electrostatics are considered, standard covalent interaction terms). The
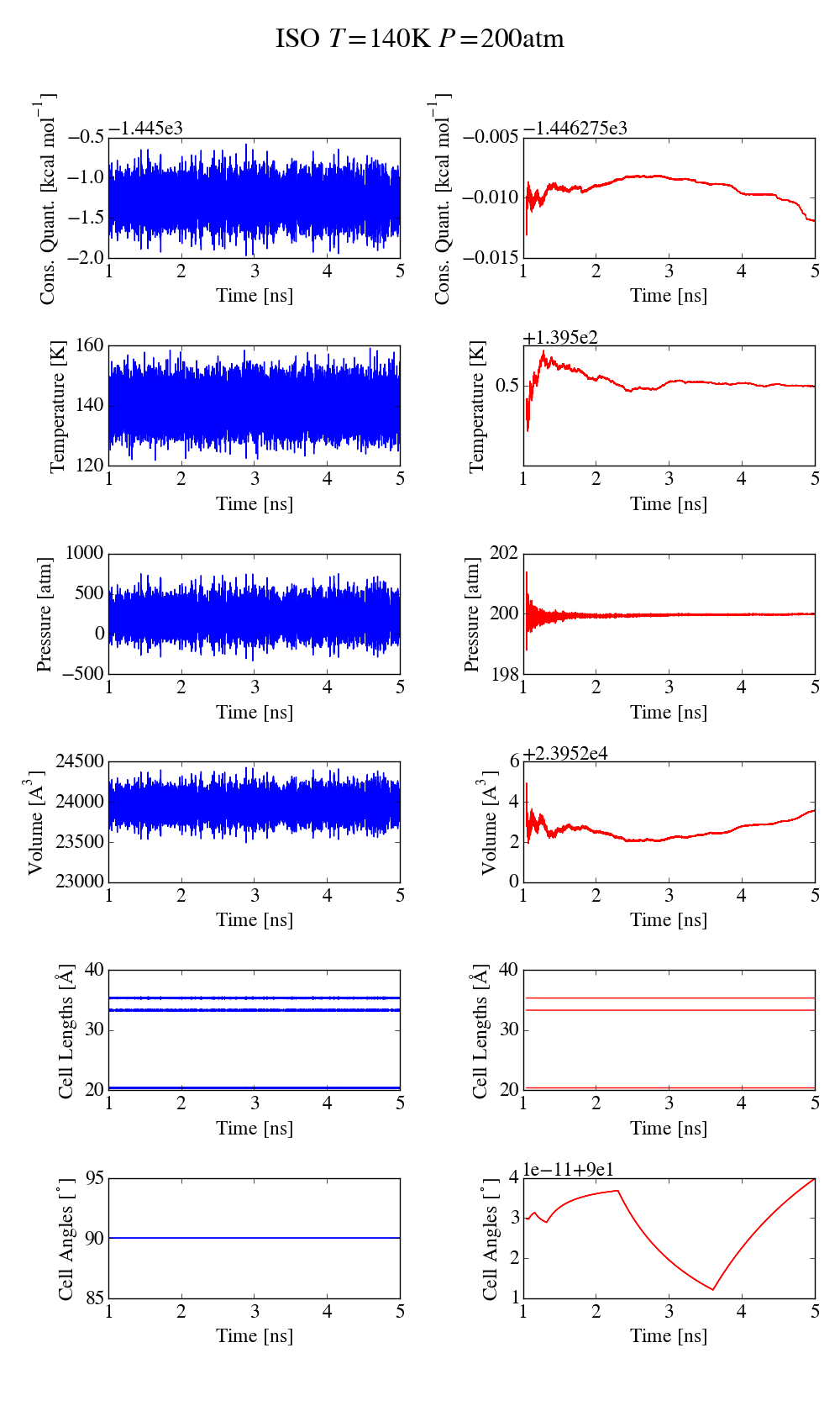
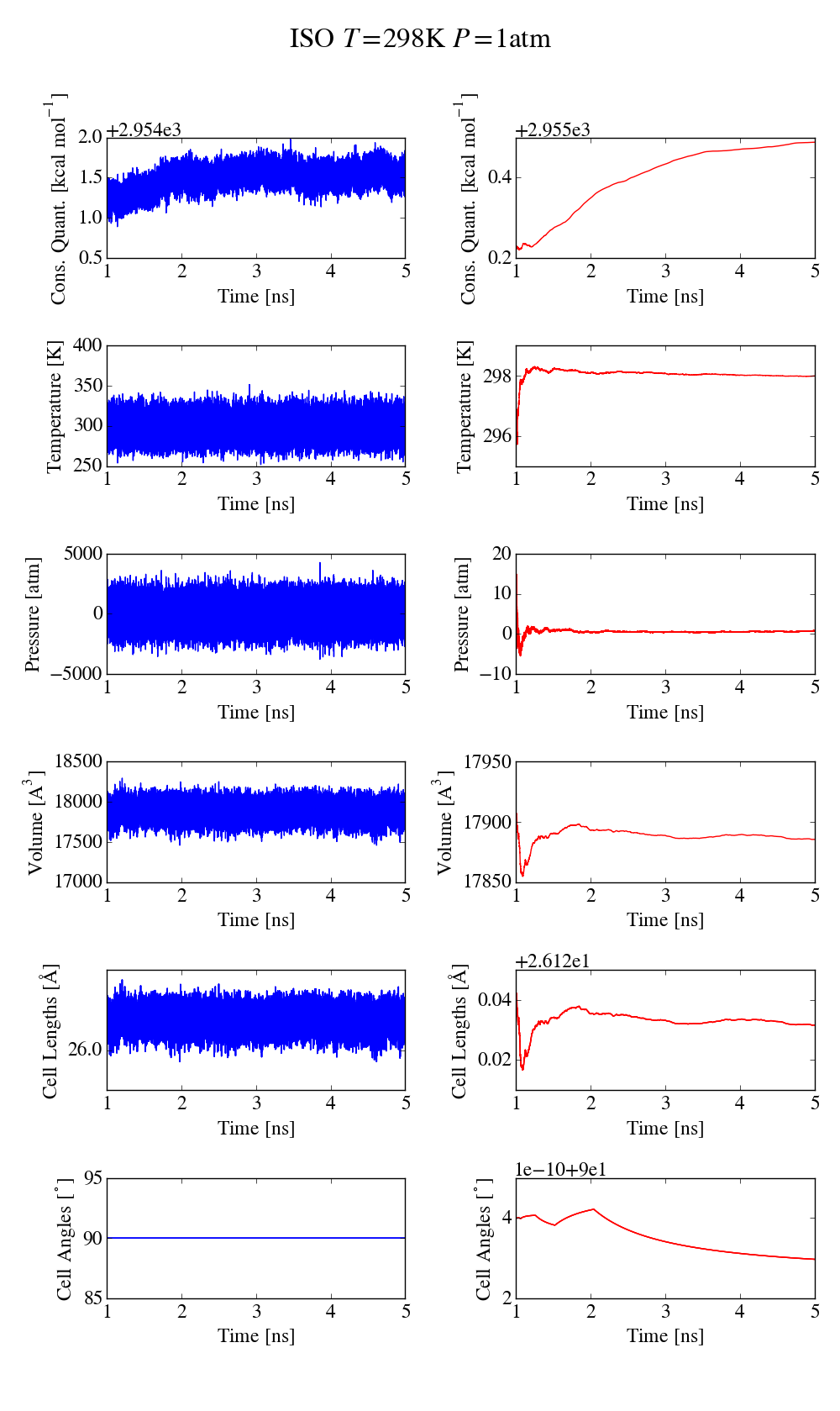
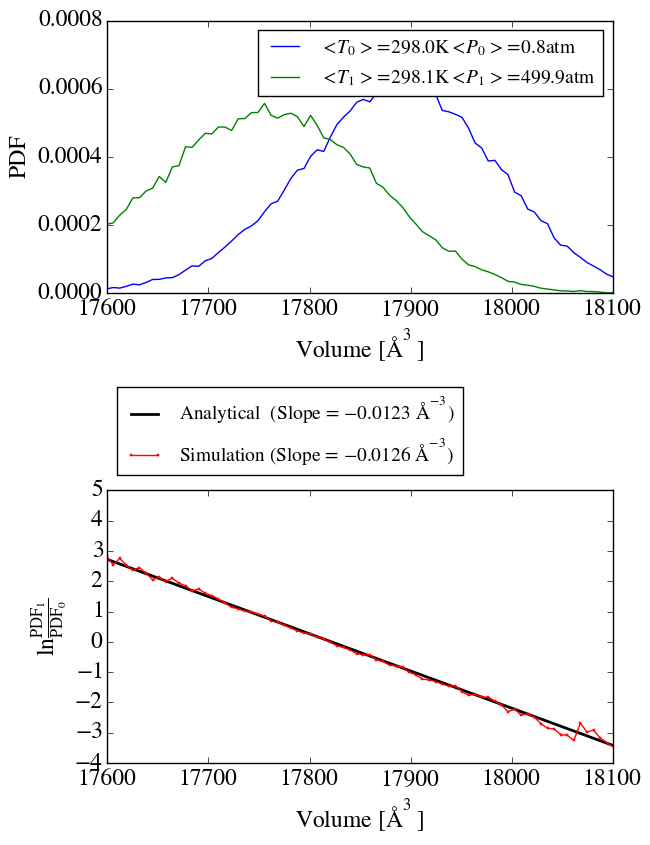
first simulation is done at 298K and an isotropic pressure of 1atm, with
the iso keyword to only allow isotropic fluctuations, with 1ns
equilibration and 4ns for data collection (time step 0.5ns). The plot
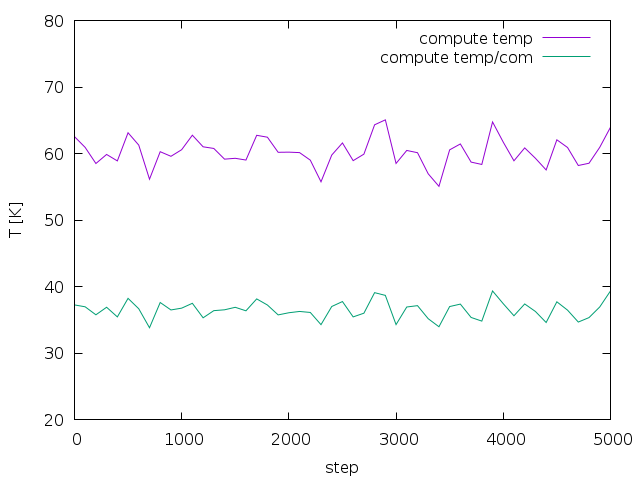
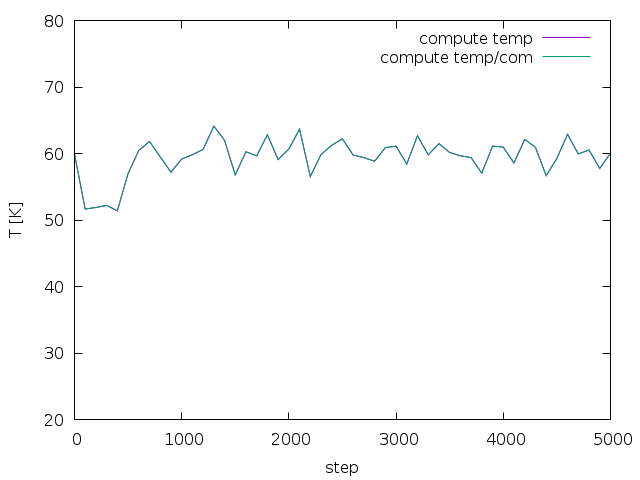
md_iso_t0-p0.png shows some quantities during the MD run in blue and the
corresponding running averages in red. Everything seems fine and there
is no systematic drift in the conserved quantity. I also performed a
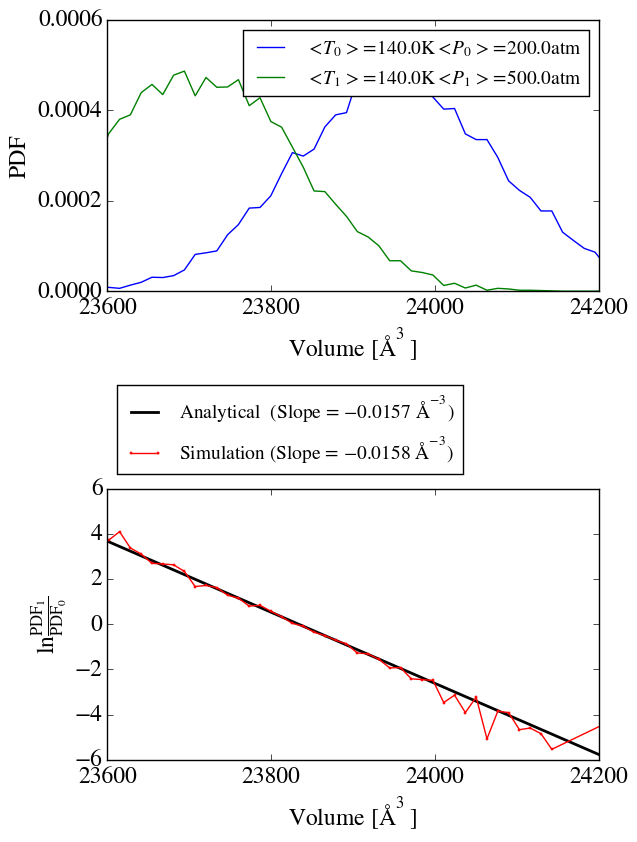
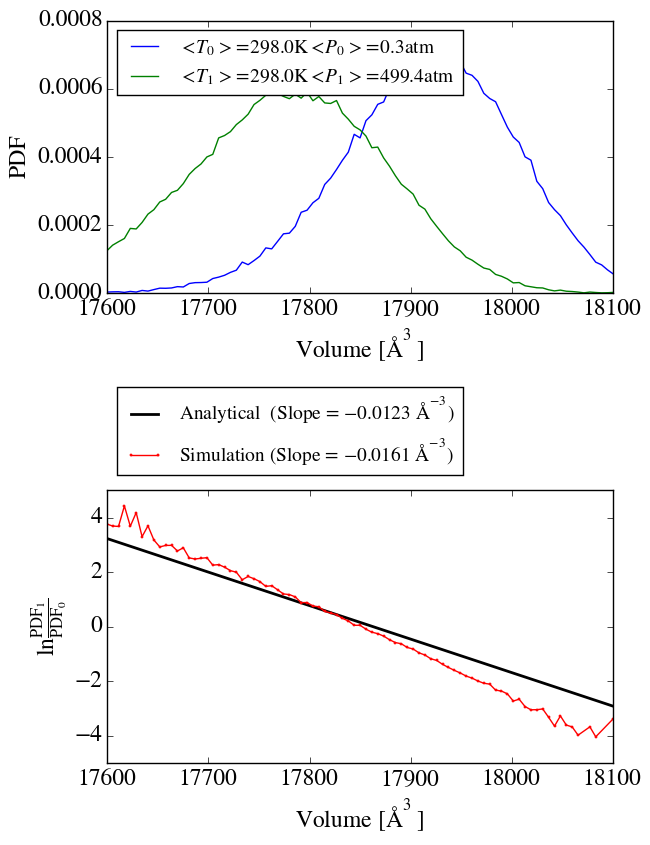
second simulation at a pressure of 500atm and applied the method of
Shirts (10.1021/ct300688p) to check the ensemble sampling. As shown in
shirts_iso_press.png, the analytical slope corresponds to the calculated
one from the simulation. Up to know everything seems to work perfectly.
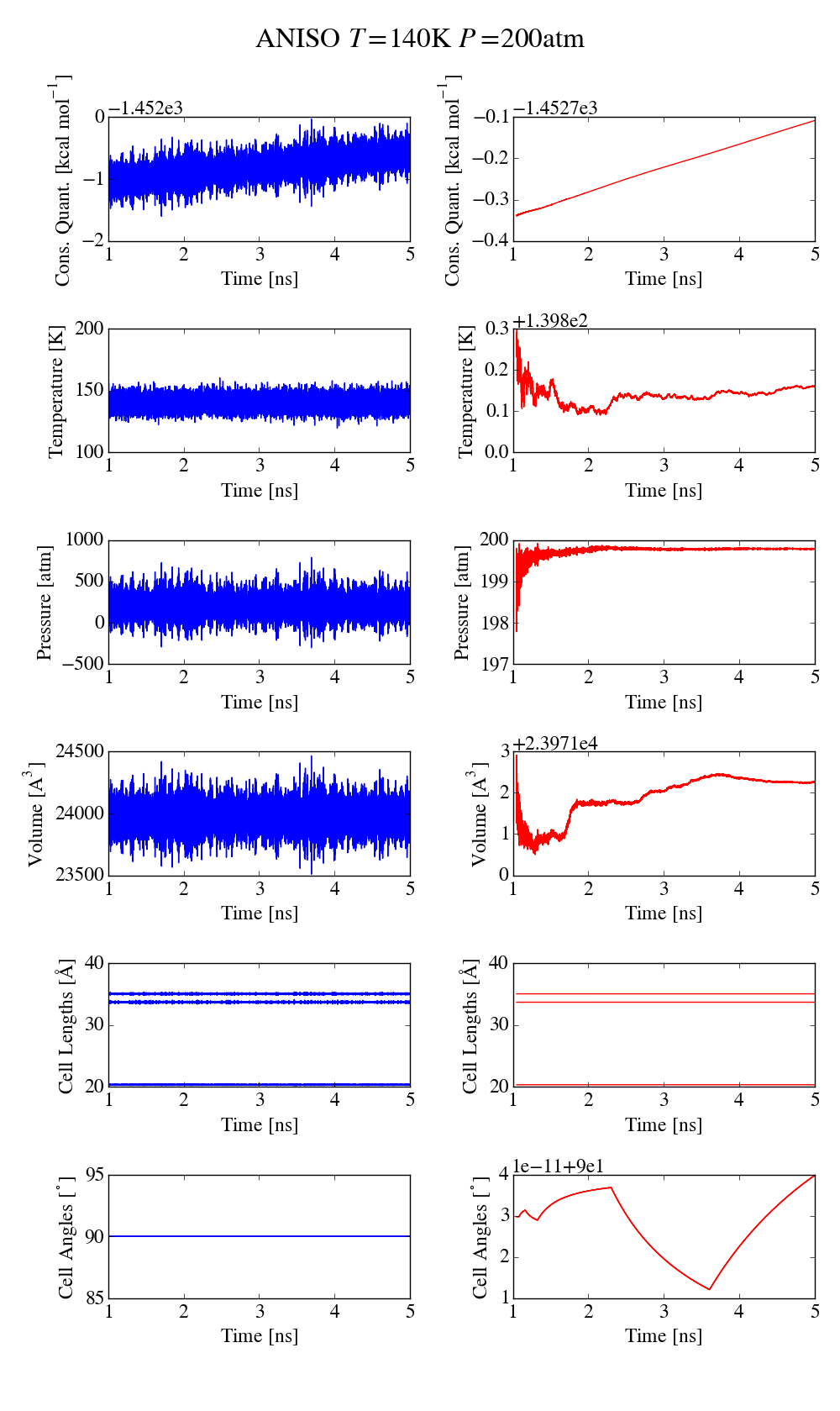
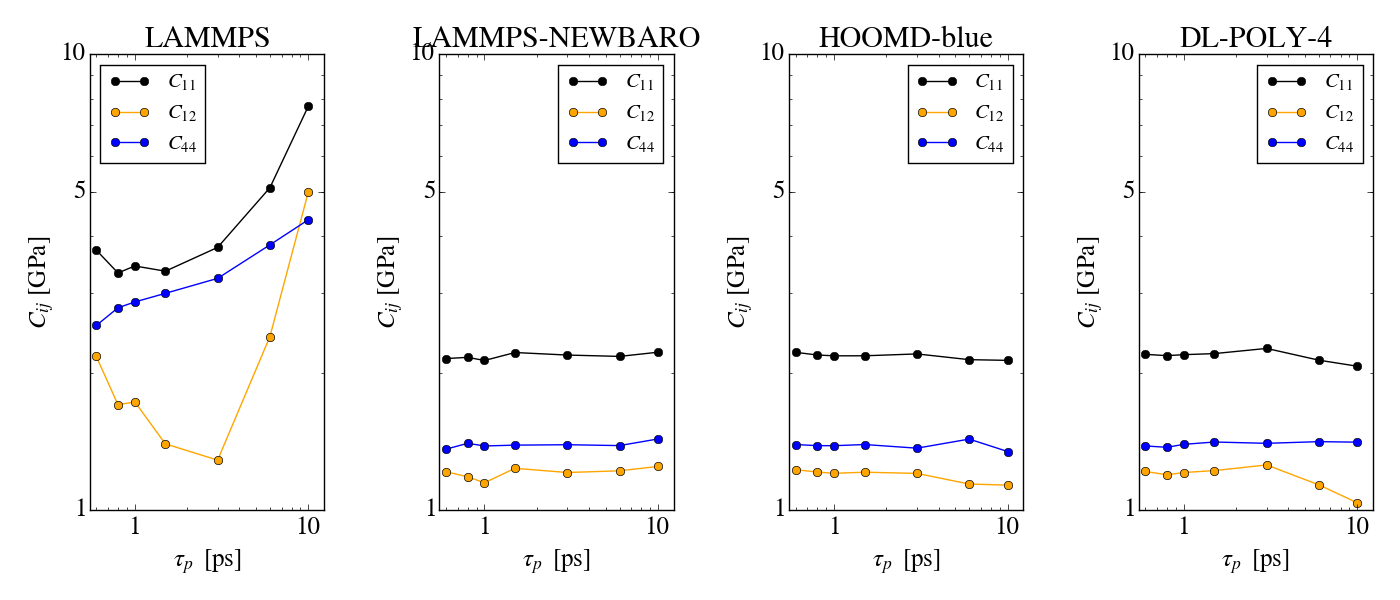
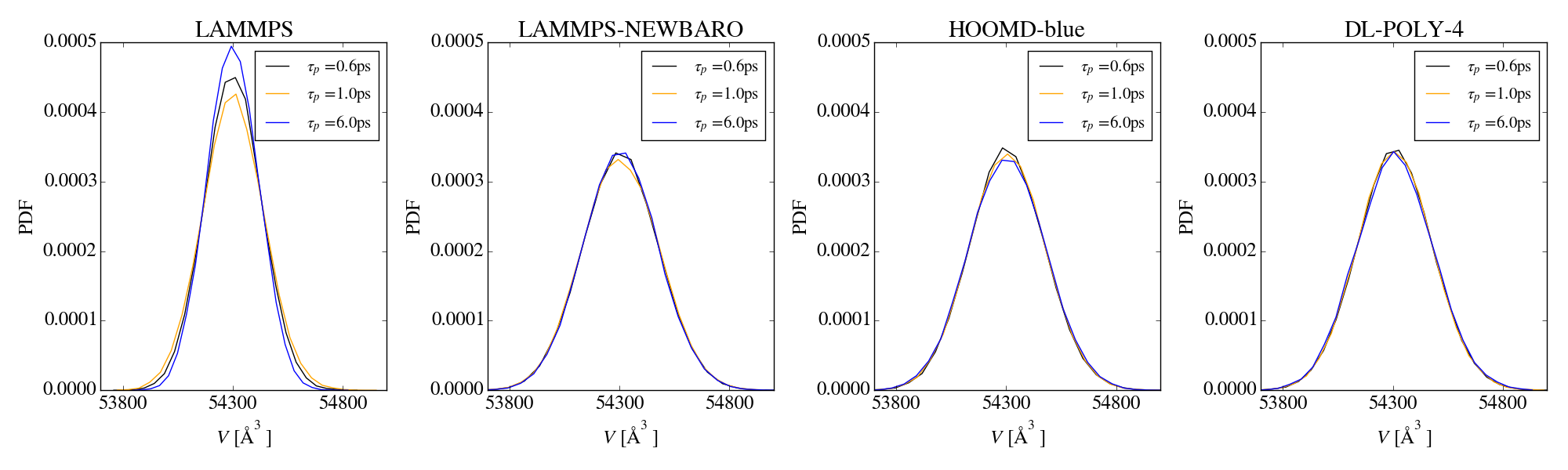
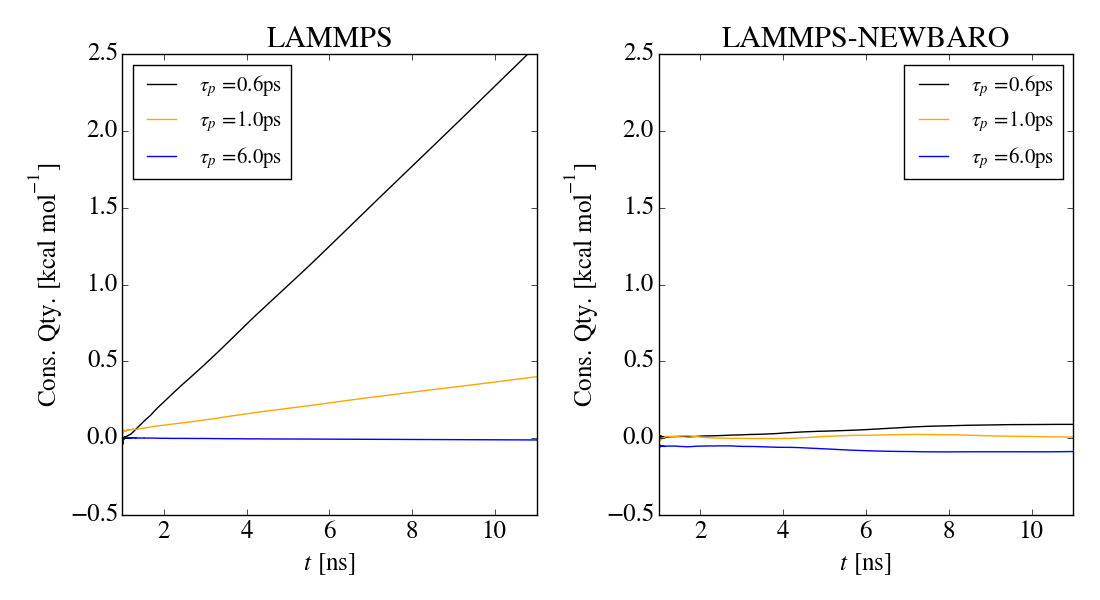
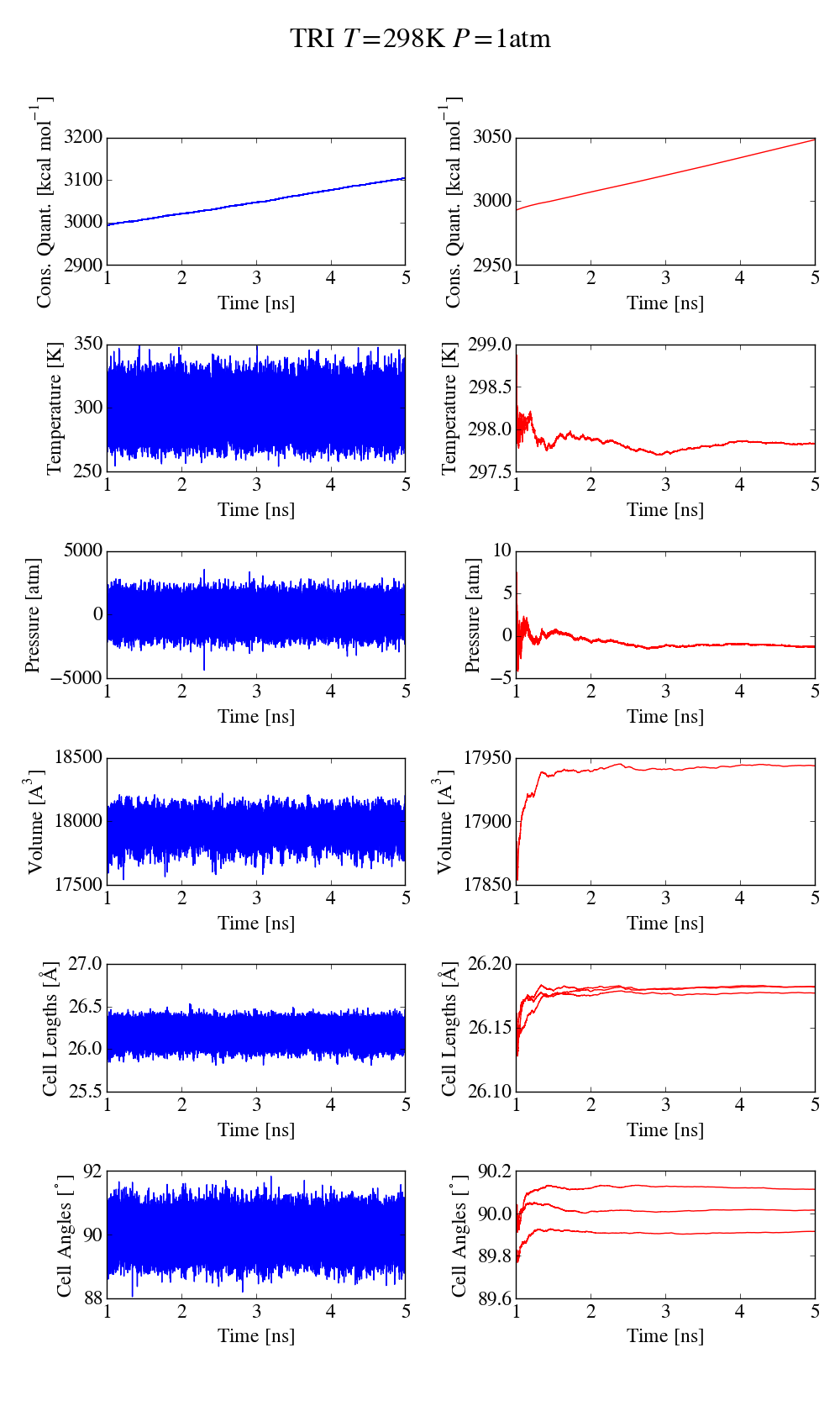
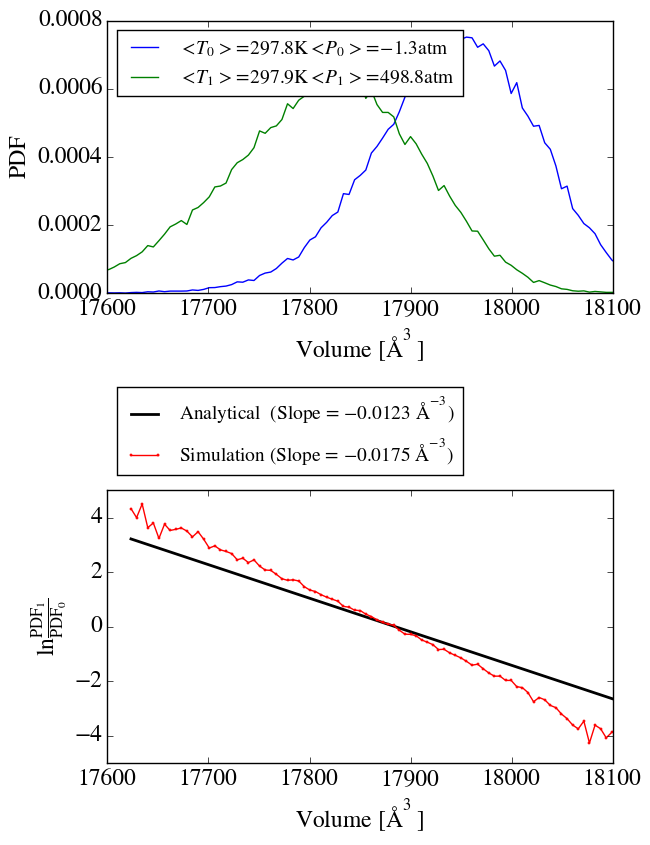
Things go haywire when allowing anisotropic cell fluctuations (using for
instance the aniso or tri keywords). The plots of the same simulations
as before (where only the iso keyword is changed, the applied pressure
is still isotropic) show that there is a clear drift in the conserved
quantity (5kcal/mol/ns for aniso, more than 10kcal/mol/ns for tri).
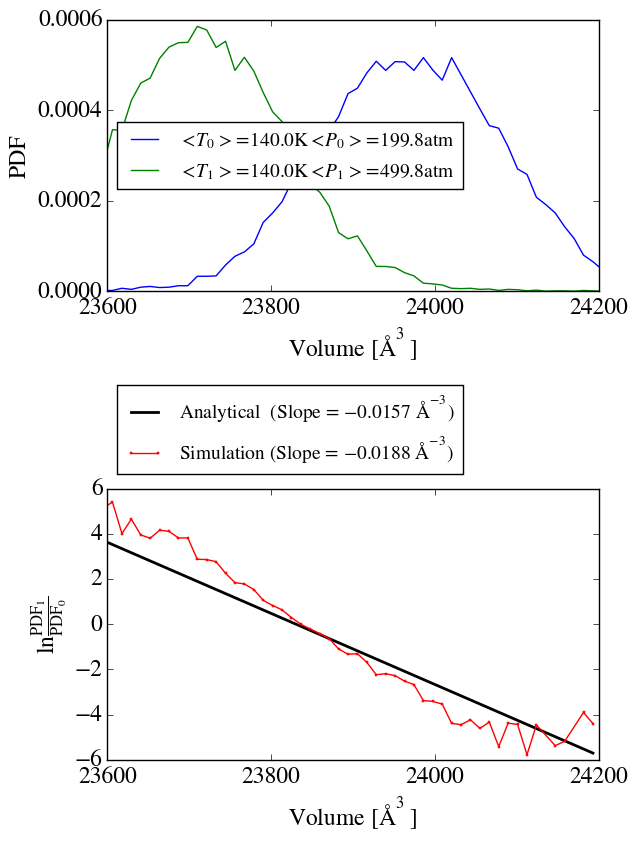
Additionally, by applying the method of Shirts I see a significant
deviation between the analytical and simulated ratio of volume
distributions, which indicates that the wrong ensemble is sampled.
I have tried changing most of the sampling settings (timestep, damping
parameters of thermo/barostat, number of thermostat chains for
particles/barostat particles, using the mtk term or not), but the
conclusions are always the same: for isotropic cell fluctuations
everything seems fine, but things go wrong as soon as anisotropic cell
fluctuations are allowed.
I have looked into the source code, but could not pinpoint the problem.
Some possible problems I encountered:
1) The integration scheme by Tuckerman (10.1088/0305-4470/39/19/S18),
which is said to be followed, does not provide equations for anisotropic
fluctuations. These are given by Yu (10.1016/j.chemphys.2010.02.014). A
difference between both cases is the number of degrees of freedom for
the barostat (1 for iso, 3 for aniso, 6 for tri), but it seems this is
not used in the source code.
2) A separate Nose-Hoover thermostat is used for the barostat particles,
which acts first and last in the integration scheme. I do not understand
fullly why the Liouville operator is written in that way.
3) The derivations by Tuckerman always assume symmetric cell tensors.
The LAMMPS convention is however different, perhaps this leads to some
changes? (Although this would not explain the problems with the aniso
case, where for the test system the cell tensor is diagonal).
Lammps version is from the master branch on 07/03/2018
Does anybody have any suggestions how the mentioned problems could be
solved?
Regards
Steven Vandenbrande

data.system (171 KB)
in.system.aniso (1.98 KB)
in.system.iso (1.98 KB)
in.system.tri (1.98 KB)