hi lammps users,
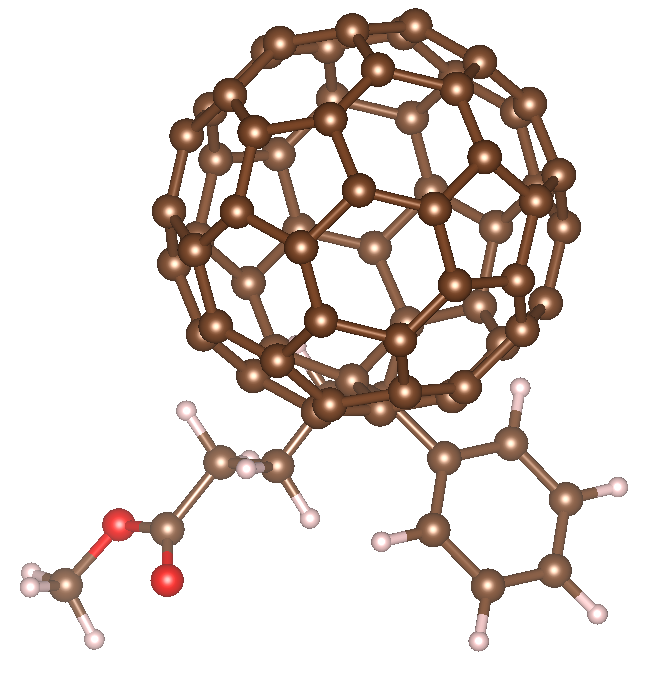
I am modeling the PCBM system, which is a C60 (atom-type 1, atom-id 1-60) and a 28 atom “satellite” molecule (atom-type 2-11, atom-id 61-88), see attached pcbm.png.
The atom-type 1, atom-id 1-60 I want to model using the Tersoff potential.
The atom-type 1, atom-id 1,2 are bonded to atoms in the “satellite” molecule through harmonic bonds:
16 7 61 1
17 7 61 2
3-body angle harmonic bonds:
22 10 1 61 70
23 10 2 61 70
24 11 1 61 2
47 19 1 61 70
48 19 2 61 70
49 20 1 61 62
50 20 2 61 62
55 25 2 1 61
56 25 6 1 61
57 25 9 1 61
58 25 1 2 61
59 25 3 2 61
60 25 12 2 61
ad 4-body dihedral bonds:
1 20 1 61 70 71
2 20 2 61 70 71
1 21 1 61 70 71
2 21 2 61 70 71
1 22 1 61 70 71
2 22 2 61 70 71
1 23 1 61 70 80
2 23 1 61 70 81
3 23 2 61 70 80
4 23 2 61 70 81
1 24 2 1 61 62
2 24 6 1 61 62
3 24 9 1 61 62
4 24 1 2 61 62
5 24 3 2 61 62
6 24 12 2 61 62
1 25 2 1 61 70
2 25 6 1 61 70
3 25 9 1 61 70
4 25 1 2 61 70
5 25 3 2 61 70
6 25 12 2 61 70
I see that the special_bonds command does not support Tersoff:
http://lammps.sandia.gov/doc/special_bonds.html
"
Is there anyway to enforce the Tersoff interaction between all atom-type 1 while also retaining the bonding term with the satellite molecule without significantly changing my input structure format?
When I run the attached structure and script the C60 atoms “explode”. I’m guessing this because either:
a) the Tersoff potential is not being applied to atom-type 1, atom-id 1-60 ecause these atoms are part of molecule-id 1.
or
b) the non-bonded lj/cut and/or coul/wolf are turned on between atom-type 1, atom-id 1-60.
I still need the non-bonded (lj/cut and coul/wolf) interactions to apply to intra-C60 atoms, so that I could simulate crystalline C60 for example.
If I cannot keep a similar input structure, it is not obvious to me how to set the potentials up properly.
I apologize if the solution for this is obvious with more careful reading of the documentation.
Thanks,
Jason
C.tersoff (734 Bytes)
fort.26 (13.4 KB)
lmp.in.anneal (5.89 KB)