Dear Lammps users,
I am using the Lammps version “3 Aug 2016-ICMS”
I am facing a problem while simulating the core-shell model of PbTiO3,
whose potentials were borrowed form:
“Sepliarsky, M., Asthagiri, A., Phillpot, S. R., Stachiotti, M. G. & Migoni, R. L. Atomic-level simulation of ferroelectricity in oxide materials. Curr. Opin. Solid State Mater. Sci. 9, 107–113 (2005).”
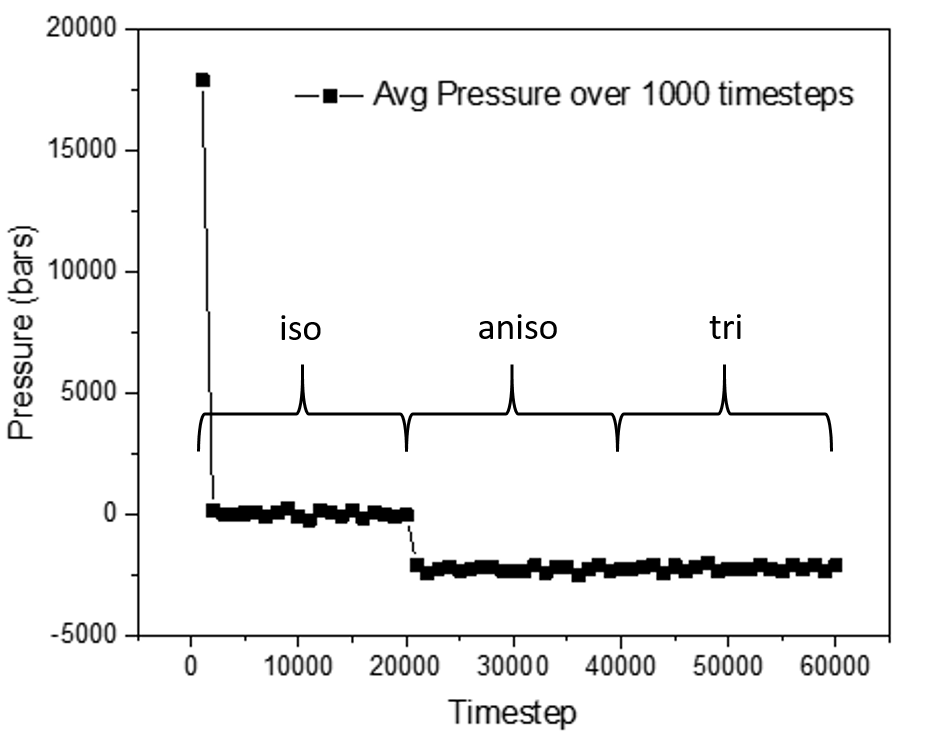
When using NPT ensemble, if I restrain pressure to be isotropic, then the input and output pressure match. However, if I set the pressure to be either anisotropic or triclinic, then the output pressure deviates substantially, and consistently settles down at a value other than that given in the input. I have verified that the number of atoms in the box does not affect this anomaly.
I have attached the input file, the data file, and a graph showing the difference in pressure using the three different pressure restraints.
Please suggest what is going wrong here.
Thanks and Regards,
Badari
PTO.in (2.04 KB)
PTO.xyz (33.7 KB)