Dear LAMMPS users and developer team,
I am trying to do structure relaxation of LiFePO4 using Lammmps (10
Feb 2015 version). I had done multiple trials with different set of
lattice coordinates and force fields. However, every time I encounter
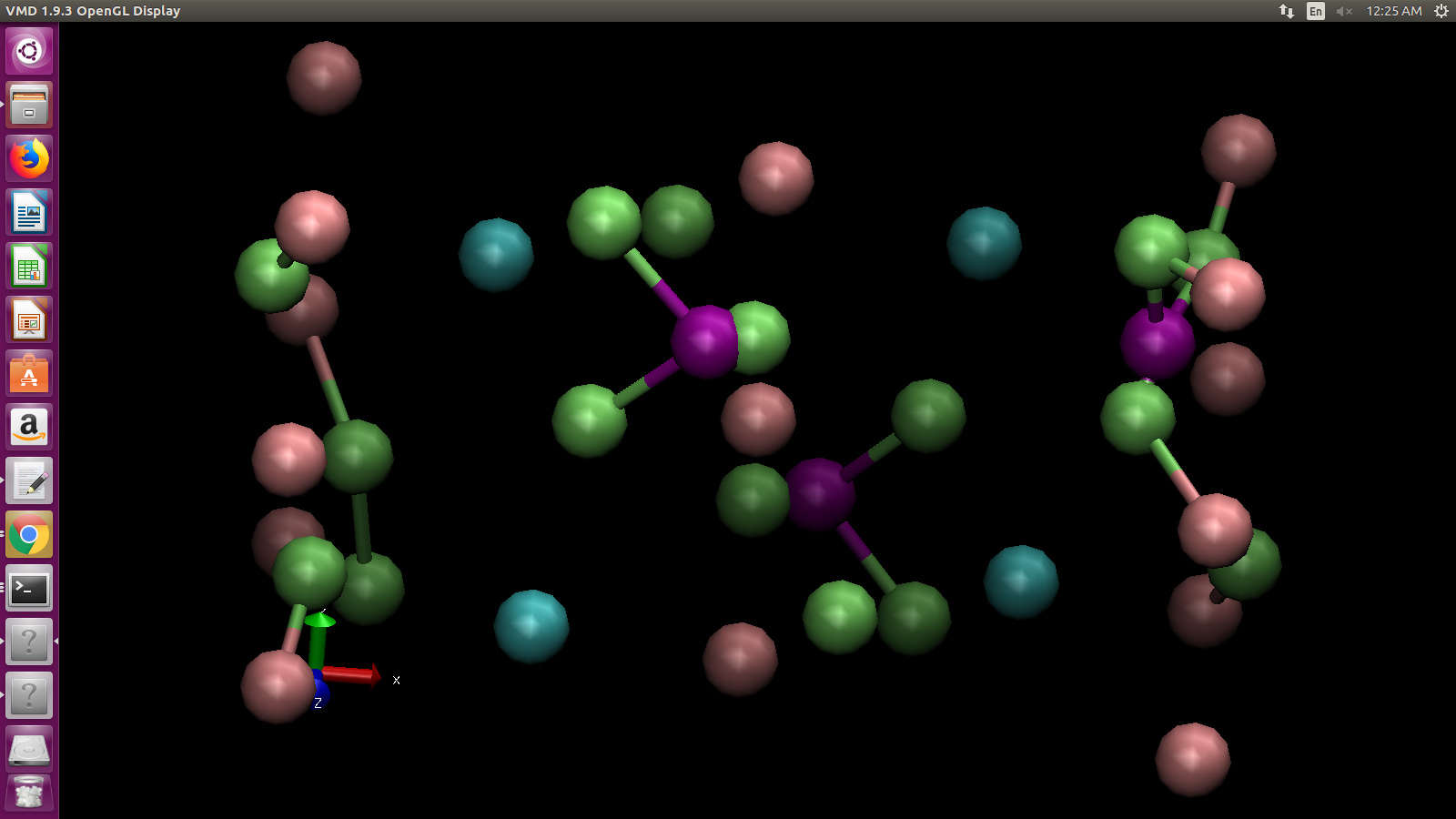
the issue of overlapping of atoms in minimised structure, when I
visualise the structure in VMD. (Image and trajectory files attached
below). From my observation and inference, the overlapping effect of
Oxygen and Phosphorous atoms is only due to Coulombic forces, there is
no effect of Buckingham force field values. I have tried varying
Buckingham pair coefficients and used various force fields. The issue
still persists. Please let me know if I am missing anything in the
Lammps input script or making any mistake in syntax. Thanking you in
advance.
The script in lammps input file is as follows:
############INITIALIZATION#####################
units metal
dimension 3
boundary p p p
atom_style charge
################ATOM DEFINITION################
region box block 0 10.06 0 5.89 0 4.65
lattice custom 1 a1 10.06 0.00000 0.00000 a2 0.0000 5.89 0.00000
a3 0.00000 0.00000 4.65 &
basis 0.00 0.00 0.00 &
basis 0.50 0.50 0.50 &
basis 0.50 0.00 0.50 &
basis 0.00 0.50 0.00 &
basis 0.28 0.25 0.98 &
basis 0.72 0.75 0.20 &
basis 0.78 0.25 0.52 &
basis 0.22 0.75 0.48 &
basis 0.09 0.25 0.42 &
basis 0.91 0.75 0.58 &
basis 0.59 0.25 0.08 &
basis 0.41 0.75 0.92 &
basis 0.09 0.25 0.75 &
basis 0.45 0.25 0.21 &
basis 0.17 0.04 0.29 &
basis 0.91 0.75 0.25 &
basis 0.55 0.75 0.79 &
basis 0.83 0.96 0.71 &
basis 0.59 0.25 0.75 &
basis 0.41 0.75 0.25 &
basis 0.95 0.25 0.29 &
basis 0.05 0.75 0.71 &
basis 0.67 0.46 0.21 &
basis 0.83 0.54 0.71 &
basis 0.33 0.96 0.79 &
basis 0.33 0.54 0.79 &
basis 0.14 0.46 0.29 &
basis 0.67 0.04 0.21
create_box 4 box #4 is number of types atoms
create_atoms 4 region box &
basis 1 1 &
basis 2 1 &
basis 3 1 &
basis 4 1 &
basis 6 2 &
basis 7 2 &
basis 5 2 &
basis 8 2 &
basis 9 3 &
basis 10 3 &
basis 11 3 &
basis 12 3 &
basis 13 4 &
basis 14 4 &
basis 15 4 &
basis 16 4 &
basis 17 4 &
basis 18 4 &
basis 19 4 &
basis 20 4 &
basis 21 4 &
basis 22 4 &
basis 23 4 &
basis 24 4 &
basis 25 4 &
basis 26 4 &
basis 27 4 &
basis 28 4
mass 1 6.941 #Li
mass 2 55.845 #Fe
mass 3 30.97 #P
mass 4 16.00 #O
set type 1 charge +1
set type 2 charge +2
set type 3 charge +5
set type 4 charge -2
#neigh_modify delay 0 every 1 check no
###############SETTINGS#######################
kspace_style ewald 1.0e-4
pair_style buck/coul/long 10
pair_coeff 1 4 632.1018 0.2906 0.0
pair_coeff 2 4 1105.2409 0.3106 0.0
pair_coeff 3 4 897.2648 0.3577 0.0
pair_coeff 4 4 22764.3 0.149 44.53
pair_coeff * * 0.000 1.000 0.0
minimize 1.0e-4 1.0e-6 100 1000
write_dump all custom aftermin3.lammpstrj id type x y z vx
vy vz mass q
#####################end#############################
Regards,
Saurabh
aftermin1.lammpstrj (1.31 KB)
beforemin1.lammpstrj (1.17 KB)