While using reaxff force field for Li and Si (developed by Van Duin, et al.), I initially used pair_style reax and got the expected results from my simulation.
However while using pair_style reax/c I am getting different, and quite unexpected results for the otherwise exactly same script. From the documentation I find that,
“The reax style differs from the reax/c style in the lo-level implementation details. The reax style is a Fortran library, linked to LAMMPS. The reax/c style was initially implemented as stand-alone C code and is now integrated into LAMMPS as a package.”
From this I am unable to understand why I get such drastic difference between the two pair styles implemented. This also affects any potential simulation using pair_style hybrid, as I can not use pair_style reax in that, I have to use pair_style reax/c.
Have anyone here faced similar problem, or point out to a potential reason for this?
Each of the two ReaxFF implementations in LAMMPS, reax and reax/c, comes with its own default settings for different elements of the ReaxFF potential, and each also provides options allowing the user to override those settings. Depending on the application, these differences may have no effect on the result, or result in “drastic difference.”
You should avoid assuming that the simulation that gives you the answer you like the best is actually the one that uses a faithful implementation of the original ReaxFF parameterization for that system. Instead, do rigorous testing by reproducing published results for the ReaxFF parameterization that you are trying to recreate in LAMMPS. If this is too much work for you, then you should confine your research to only those validated reax/c parameterizations that we provide with LAMMPS in examples/reax/<example_dir>.
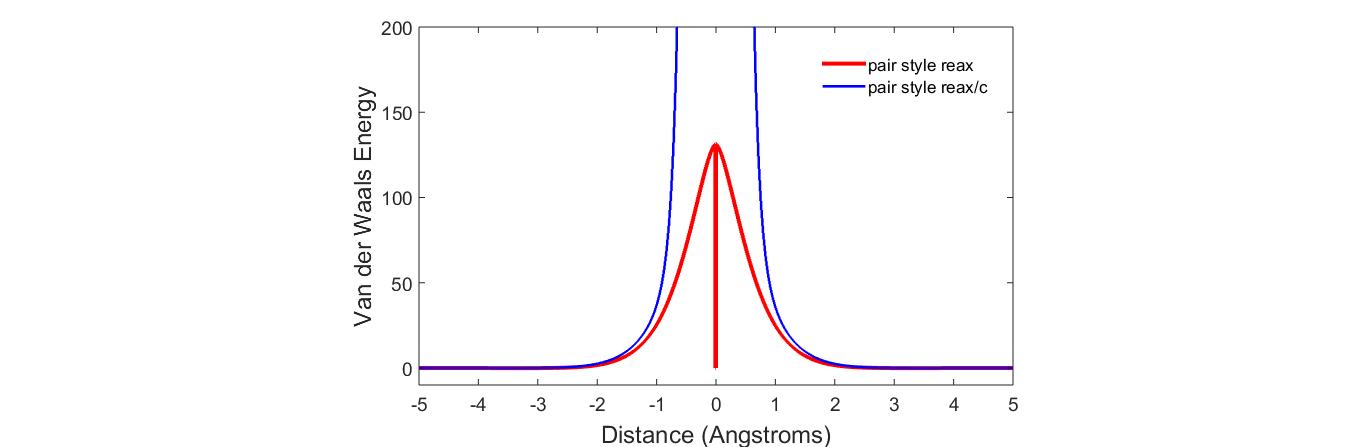
Thank you for your detailed response. I have reproduced published results for the ReaxFF parameterization using pair_style reax. However the behavior is fundamentally different when using pair style reax/c. To capture this difference I also plotted the potential energy vs distance curves for the same parameterization, using the two different commands- pair_style reax, and pair_style reax/c. I have attached a slide containing the commands and the difference in the results I obtained…
Thank you for your detailed response. I have reproduced published results
for the ReaxFF parameterization using pair_style reax. However the
behavior is fundamentally different when using pair style reax/c. To
capture this difference I also plotted the potential energy vs distance
curves for the same parameterization, using the two different commands- pair_style
reax, and pair_style reax/c. I have attached a slide containing the
commands and the difference in the results I obtained..
You MUST use fix qeq/reax with pair reax/c, or it'll never agree with
literature results of systems with partial charges.
Exactly. Swastik, it is very likely that you did not have the “fix qeq/reax” command for charge equilibrations. It is obvious from the potential energy curve that reax/c is missing some energy – which is most likely from Coulomb interactions. If you did not have fix qeq/reax and your charges were all zero, you would be missing the electrostatics.
While using reaxff force field for Li and Si (developed by Van Duin, et al.), by using pair_style reax/c I am getting different results for potential energy against distance, as well as for force against distance, than the results I obtain using pair_style reax, specially when the distance between the atoms is low. I have attached the results in the accompanying slides. When using pair_style reax/c I have also used the “fix qeq/reax” command for charge equilibrations. I am looking forward to any suggestion regarding what might be a reason for this?
This is a good way to test the different definitions of ReaxFF in the two pair_styles.
The match is good around the equilibrium separation. The differences seem to occur at short separations and at large separations. In order to isolate the causes of these differences, you should compare the different components of the energy. See the examples/reax scripts for more information on how to do this. Once you know where the difference is coming from, you will have to figure out which of the two is faithful to the original ReaxFF Si-Li model (if either). If you want to use pair_style reax/c (recommended) and it is not faithful, you may be able to change it using the control file settings (see documentation). It is possible that you will need to change something in the code to achieve the correct Li-Si potential.
Hi! Thank you so much for the suggestions. I checked the different components of the energy for Li-Li interaction, and I see a huge difference in the Van der Waals contribution for reax and reax/c implementation. It seems like it stems from lack of shielding implementation in repulsion in reax/c, but the warning message I get from reax/c implementation is this:
“Warning: inconsistent vdWaals-parameters
Force field parameters for element X
indicate shielding without inner wall, but earlier
atoms indicate different vdWaals-method.
This may cause division-by-zero errors.
Keeping vdWaals-setting for earlier atoms.
Warning: changed valency_val to valency_boc for X
Warning: inconsistent vdWaals-parameters
Force field parameters for element X
indicate shielding without inner wall, but earlier
atoms indicate different vdWaals-method.
This may cause division-by-zero errors.
Keeping vdWaals-setting for earlier atoms.
Warning: changed valency_val to valency_boc for X”
So this is not the warning that comes from ‘no shielding’ but rather from ‘shielding without inner wall’.
You mentioned “Once you know where the difference is coming from, you will have to figure out which of the two is faithful to the original ReaxFF Si-Li model (if either). If you want to use pair_style reax/c (recommended) and it is not faithful, you may be able to change it using the control file settings (see documentation). It is possible that you will need to change something in the code to achieve the correct Li-Si potential.”
I am trying to explore this, but I want to report this to you for help regarding the interpretation of this problem.
Just an edit to the previous mail… it is not necessary to worry about the warning for element X as I am using elements Li and Si from the potential file. But the rest of the query remains…