Dear developers of LAMMPS,
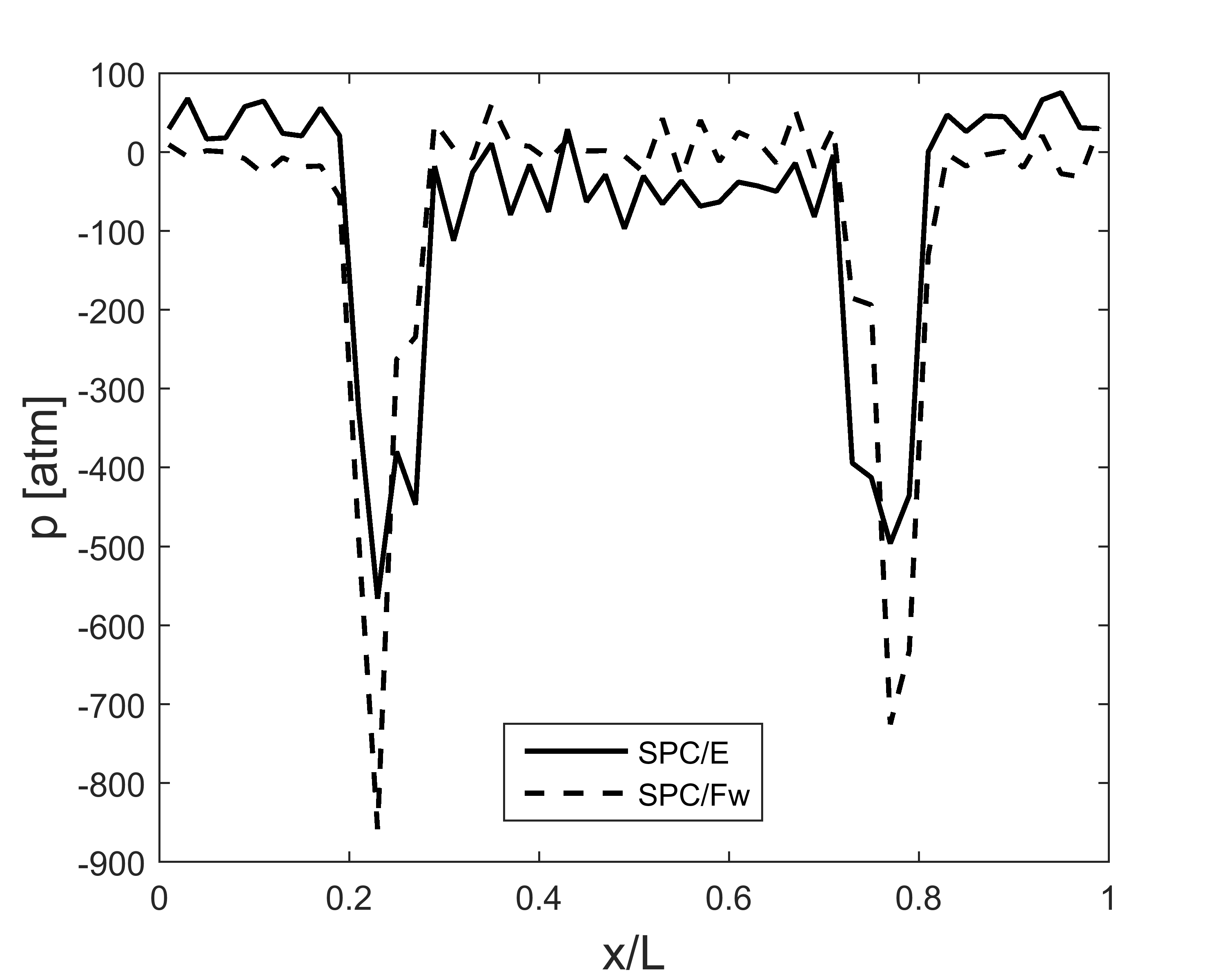
These days I am using a system as shown below to explore water-decane interaction. I apply OPLS-AA force field to decane, use large cut off for LJ pair potential instead of applying tail correction, use compute stress/atom to calculate local pressure, and average pressure curve along horizontal direction from 0.5 ns to 1 ns. The input file is shown at the bottom (remove some output lines for brevity), and a figure plotting the pressure curve along horizontal direction is shown in the attachment. I find that bulk water pressure is always about 85-100 atm higher than decane pressure if I use rigid water models such as SPC/E. For flexible water models such as SPC/Fw, this pressure difference is reduced but still exists (bulk water pressure is about 15-25 atm lower than decane pressure). I have tried to change simulation box size, LJ cutoff, run time, and shake tolerance, but the difference could not be apparently reduced.
Dear Steve,
Thank you very much for your reply. But I think I haven’t expressed my idea clearly in last email. What I study is a multicomponent multiphase problem. And I use a system like this:
To summarize, the observed pressure differences between the interiors of the two phases are :
Timestep (fs) SHAKE deltaP (atm)
1.0 yes 90
1.0 yes 30
0.5 no -20
0.5 no -10
You did not provide error bars, but I will assume these numbers are statistically distinct from 0. This is not too surprising. In a non-homogeneous system, especially one where local dynamics are coupled to global properties (kinetic energy, virial), you are always going to see subtle things like this, if you look carefully enough, which you did. Well done. The best thing would be to use fix nve and fix langevin, so that there are no constraints coupled to global properties. However, you might also want to take a step back and ask “What am I really trying to simulate?” In an inhomogeneous system, pressure is always a somewhat ill-defined quantity.
One more detail: your tdamp and pdamp values are very large. It is possible that your temperature profile is also not uniform.
Dear Aidan,
Thank you very much. I really appreciate your help.
I want to study some surface forces’s effect on wettability in nanoscale. To make my work recognized by researchers specialized in mechanics, I’d better validate Young-Laplace equation first. I would be seriously judged if I could not reduce the unphysical pressure difference. That’s why I care about this.
Yesterday I tried fix nve and fix langevin as you suggested, but the pressure profile becomes very strange (highly fluctuated and SPC/E water pressure is 200 atm lower than decane). Maybe I should run a period of npt to relax the simulation box before nve. And I plot the temperature profile and it is truly not uniform. I reduce Tdump and Pdump, and temperature profile becomes more uniform though there is no much difference to pressure profile.
I think flexible water & small time step & small Tdump have reached good enough results. If I reduce time step until no much variation occurs, could I believe my results? Is there any subtle things could not be reduced by small time step?
Looking forward to your reply.
Dear Steve,
Thank you very much for your reply. According to Young-Laplace equation, pressure difference between two phases should be zero if interface curvature is zero. That’s why I doubt my results. And I want to reduce such unphysical pressure difference because I want my work recognized by researchers specialized in mechanics.
Best wishes,
Huanhuan Tian
Dear Aidan,
I have tried to enlarge my simulation box before, but that pressure difference could not be reduced. Actually, I find the interface thickness is no larger than 2 nm. I think my system is large enough now. Based on this premise, maybe small time step could lead to convincing results?
Best wishes,
Huanhuan Tian
Dear Aidan,
I find that the method I calculated temperature curve before is not accurate. Actually temperature variation is already within 1 K for Tdump=100, Pdump=200.
Best wishes,
Huanhuan Tian