Hello Folk,
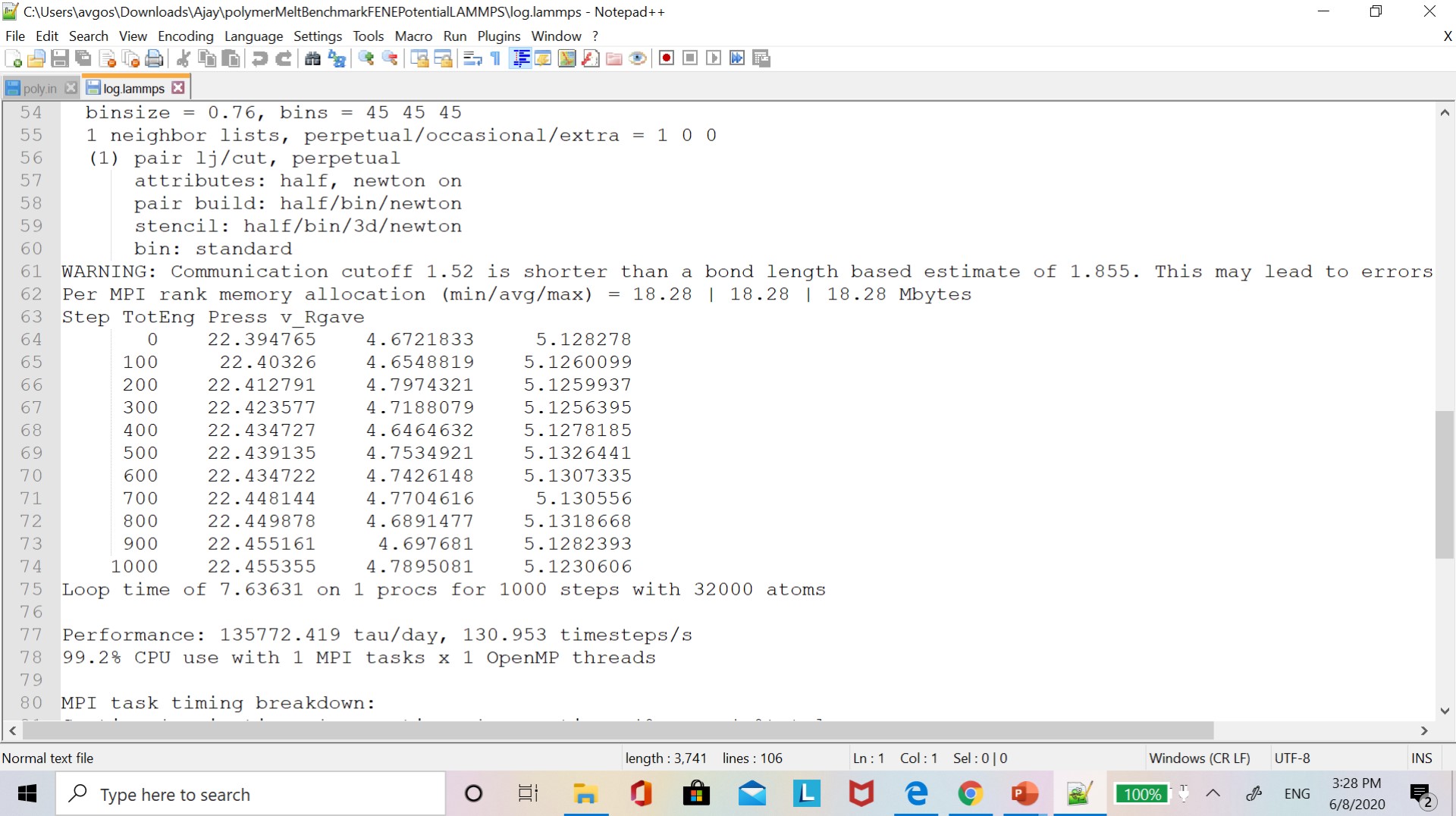
Currently, I am running a polymer melt benchmark simulation. but I am not getting the correct values of the average radius of gyration. I am attaching the input script and data file which is available in the LAMMPS benchmark study folder. I am also attaching a log file where I save radius of gyration values. I am trying to compare it with the values given in the LAMMPS manual.
this is the information available on the LAMMPS website.
Bead-spring polymer melt with 100-mer chains and FENE bonds:
- 32,000 atoms for 100 timesteps
- reduced density of 0.8442 (liquid)
- force cutoff of 2^(1/6) sigma
- neighbor skin = 0.4 sigma
- neighbors/atom = 5 (within force cutoff)
- NVE time integration
The data file is given in the LAMMPS benchmark folder for 32000 atoms. I can not attach it here due to a limitation of size less than 1 MB.
Please tell me what I am doing wrong in it.
Thanks,
Ajay

log.lammps (3.65 KB)
poly.in (664 Bytes)