Dear all,
Dear all,
Hello Kasra, just checking: are you using the unwrap option? All distances are calculated with PBCs by the colvars module, but the centers of mass of groups of atoms aren’t.
So if your colloidal particle is made by multiple molecules, you may want to check whether these are diffusing away from each other as a consequence of the biasing force.
Giacomo
Hi Giacomo,
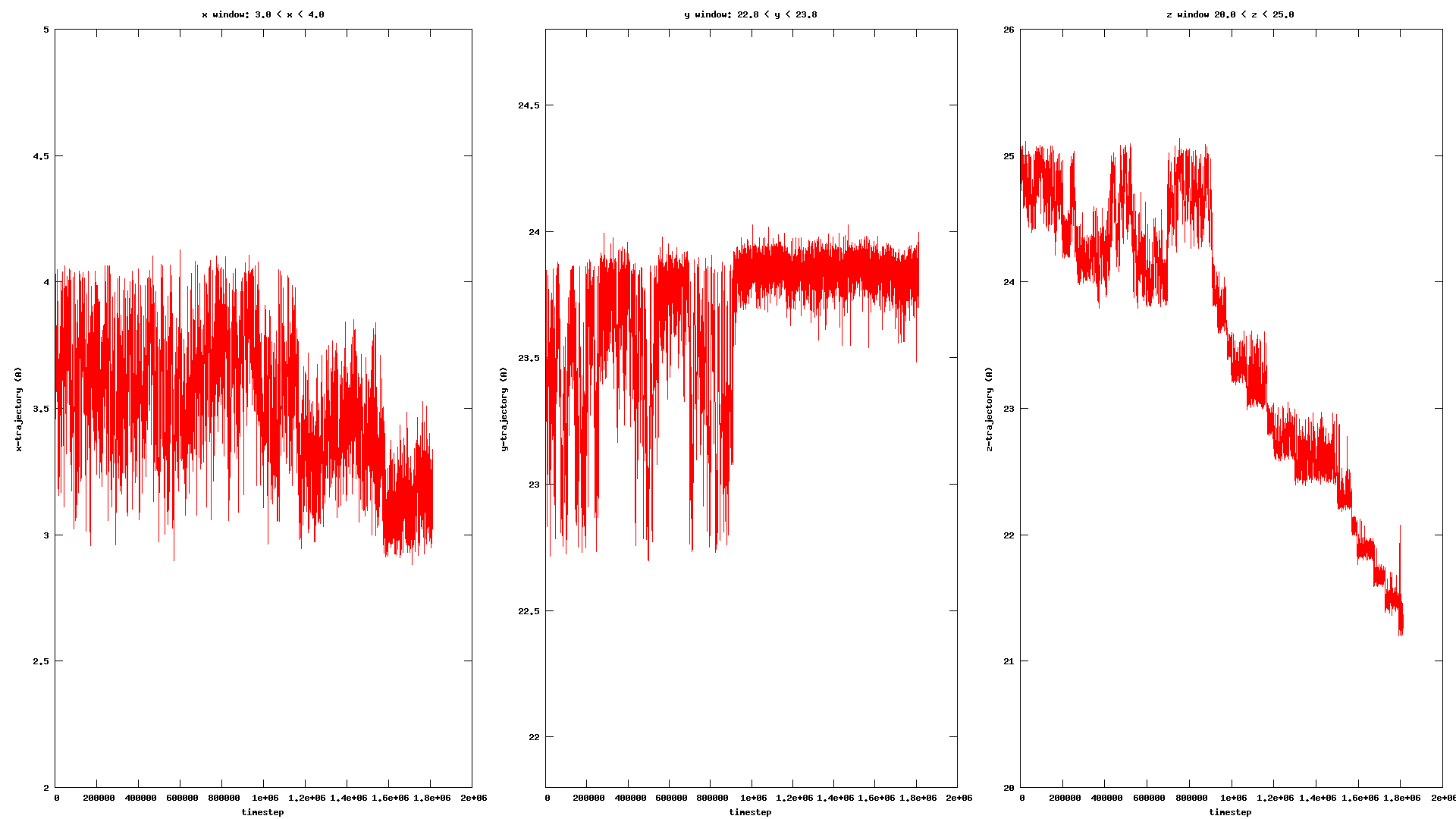
Actually, the colloidal particle that in my simulation is a single entity particle and not a group of atoms or molecules together so I’m using the default value of unwrap keyword. This happens when the Colloid is at the close proximity to the polymer wall surface, however, I don’t know how it could cause such behavior as if a wall atom is way in repulsive region of the colloid pairwise interaction, or something like that ?!
Thank you,
Kasra.
Are you calculating a center of mass of any other group of atoms?
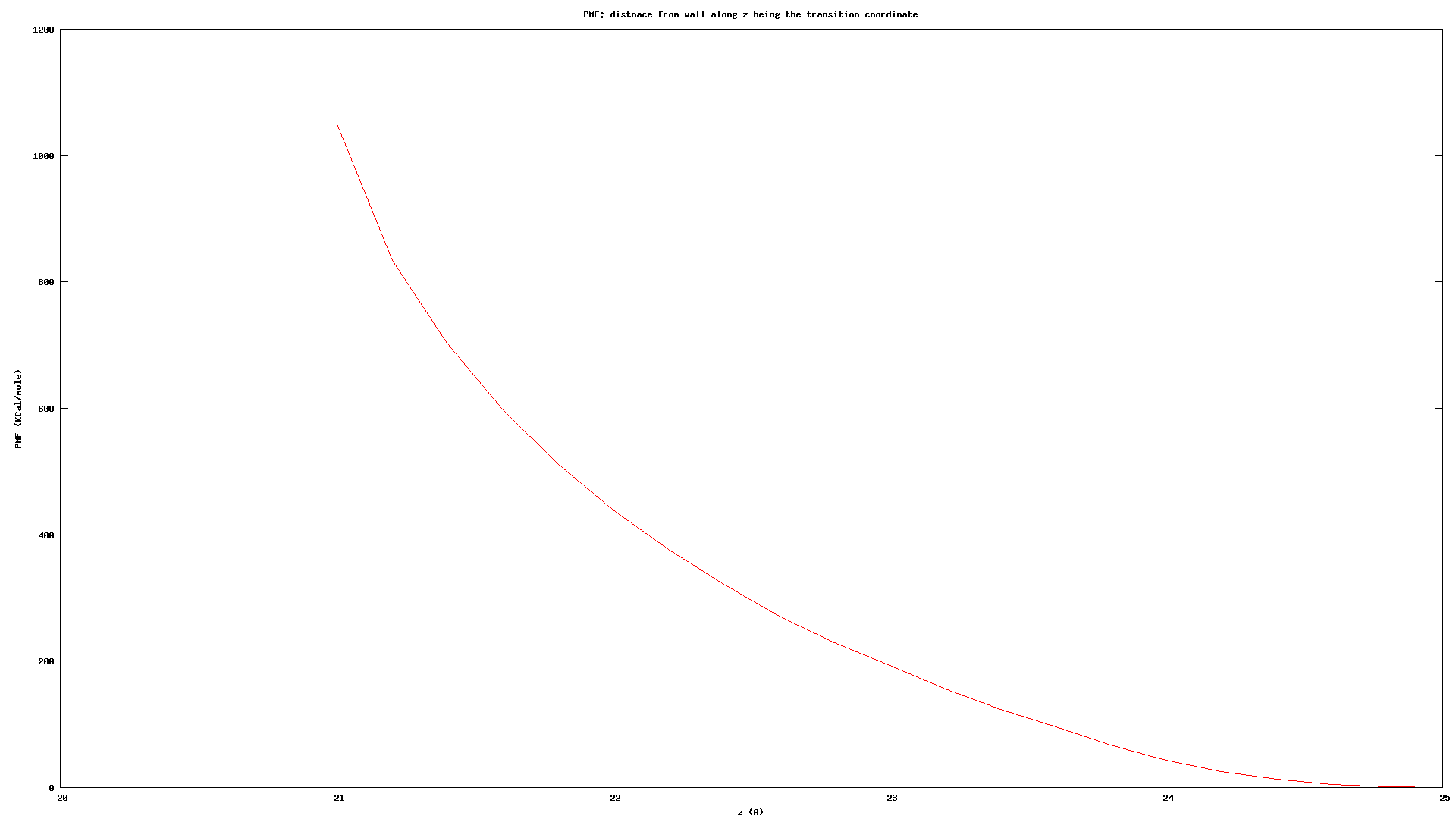
Not really, I don’t have any center of mass calculation in the LAMMPS input, I’m just tracking the colloid position which is a single entity. Also in my colvars config file, I’m using a distanceZ component for the transition coordinate (z-coordinate) using the colloid as the main and a dummyAtom at (0, 0, 0) as the reference group.
Kasra.
Because ABF accumulates the biasing force continuously, it’s hard to imagine a situation where there is a huge discontinuity caused by something other than a PBC issue.
Maybe you could do something different, then: can you pull up a configuration right before the problem occurred, keep the same collective variable definition and replace ABF with a moving harmonic restraint that moves you gradually past the point where the problem occurred.
If there was a problem with the variable’s definition, that should give you more insight in what’s going on.
Giacomo