Dear axel,
Again i’ve created with graphene single layer sheet(atomic style full) using VMD nanobuilder plugin. But, with that data file also atoms are lost.
Is their any way to correct geometry of a sheet? Can I manually change the boundaries in data file? When I tried I’m not getting good boundary conditions.
Now My new Input file is
units metal
atom_style full
boundary m p m
newton on
processors 1 1 1
dimension 3
read_data data.graphene
pair_style airebo 3.0
pair_coeff * * CH.airebo C
timestep 0.0005
#------------------- RELAXING-------------------
fix 1 all npt temp 300.0 300.0 0.05 y 0 0 0.5
thermo 2000
compute 1 all stress/atom NULL
compute 2 all pe/atom pair bond
compute 3 all reduce sum c_1[1] c_1[2] c_1[3]
thermo_style custom step temp pe ke etotal press lx ly pxx pyy c_3[1] c_3[2] c_3[3]
run 60000
unfix 1
-----------------deformation-----------------
fix 1 all nvt temp 300 300 0.05
fix 2 all ave/atom 1 1000 1000 c_1[1] c_1[2] c_1[3] c_2 fx fy fz
dump 1 all custom 1000 dump.new.* id type x y z vx vy vz c_1[1] c_1[2] c_1[3] c_2 f_2[1] f_2[2] f_2[3] f_2[4] f_2[5] f_2[6] f_2[7]
dump 2 all xyz 10000 dump.graphene.*.xyz
variable srate equal 1.0e9
variable srate1 equal “v_srate/1.0e12”
fix 3 all deform 1 y erate 0.005 units box remap x
run 600000
-> I’m first relaxing the sytem for 30ps (0.0005*60000) and then applying strain in y direction.
I’ve tried boundary conditions m and p, but the potential energy is increasing in relaxing time with fluctuating temperature and potential energy is constant when applied with strain.
Even When I try to visualize the system it is very unusual.Please have a look at the image attached.
Thanks for your co-operation Axel and have a good day ahead.
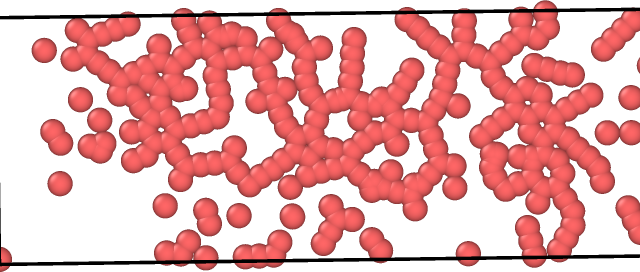
data.graphene (57.1 KB)
input.graphene (856 Bytes)