now it works fine when the “R” option in the file with the tabulated potential is used.
The working input, data, and tabulated pair potential files are in the attachment.
But I would like to avoid the initial preliminary interpolation in LAMMPS, this is why the I change the “R” option to “RSQ” in the potential.table file. But now the LAMMPS crashes badly. It starts the simulations but something strange happens with the energy and forces, they are skyrocketing and the simulations crashes during the 1st run. somehow the distance between particles gets unphysically small. The modified input (in.sq.tabulated) and table (potential.sq.table) files for which LAMMPS crashes are also attached below.
I would be grateful for any help!
What I also find strange is that the master list distance cutoff = 11, although the potential is tabulated up to 9 Angstroems.
here is how the error looks like:
$ ./lmp_ubuntu < in.sq.tabulated
LAMMPS (10 Feb 2015)
Reading data file …
orthogonal box = (0 0 0) to (24.56 24.56 160)
1 by 1 by 1 MPI processor grid
reading atoms …
2000 atoms
reading velocities …
2000 velocities
Neighbor list info …
1 neighbor list requests
update every 1 steps, delay 10 steps, check yes
master list distance cutoff = 11
Setting up run …
Memory usage per processor = 2.17587 Mbytes
Step PotEng KinEng TotEng
0 96413195 76.103586 96413271
ERROR on proc 0: Pair distance < table inner cutoff (…/pair_table.cpp:112)
in.tabulated (287 Bytes)
data.n2000 (268 KB)
potential.table (182 KB)
in.sq.tabulated (290 Bytes)
potential.sq.table (182 KB)
The error is pretty clear:
ERROR on proc 0: Pair distance < table inner cutoff (…/pair_table.cpp:112)
Two atoms are closer together than the inner cutoff, which is where
your tabulation starts (not the longer cutoff). So LAMMPS can’t
compute the force.
Steve
yes, i see it, but as I have mentioned before, when the option “RSQ” is used all quantities like the total energy are computed completely wrong, whereas with the option “R” the total energy has a correct value and the simulation does not crash. The potential is tabulated for the distances between 1 and 9 Angstroems.
Normally the atoms should not be closer as 1 Angstroem, and when I use the option “R” they are not., and everything works fine.
Instead with the “RSQ” option the simulation crashes at the 1-st step. and the precomputed value of the total energy at the 0-th step is completely wrong.
I have changed the inner cutoff from 1 to 2.5 Angstroem and now it works with the “RSQ” option!
All the needed LAMMPS script files with working parameters are attached.
The interpolation which does LAMMPS looks smooth, but unfortunately wrong. it looks like that LAMMPS shifts the abscissa (the pair distance values) (when the RSQ option is used) and then fits somehow the energy to this shifted distance values:( so that the overall pair energy output via pair_write command is shifted as is seen at the attached figure (interpolation.png).
would be very grateful for any clues!
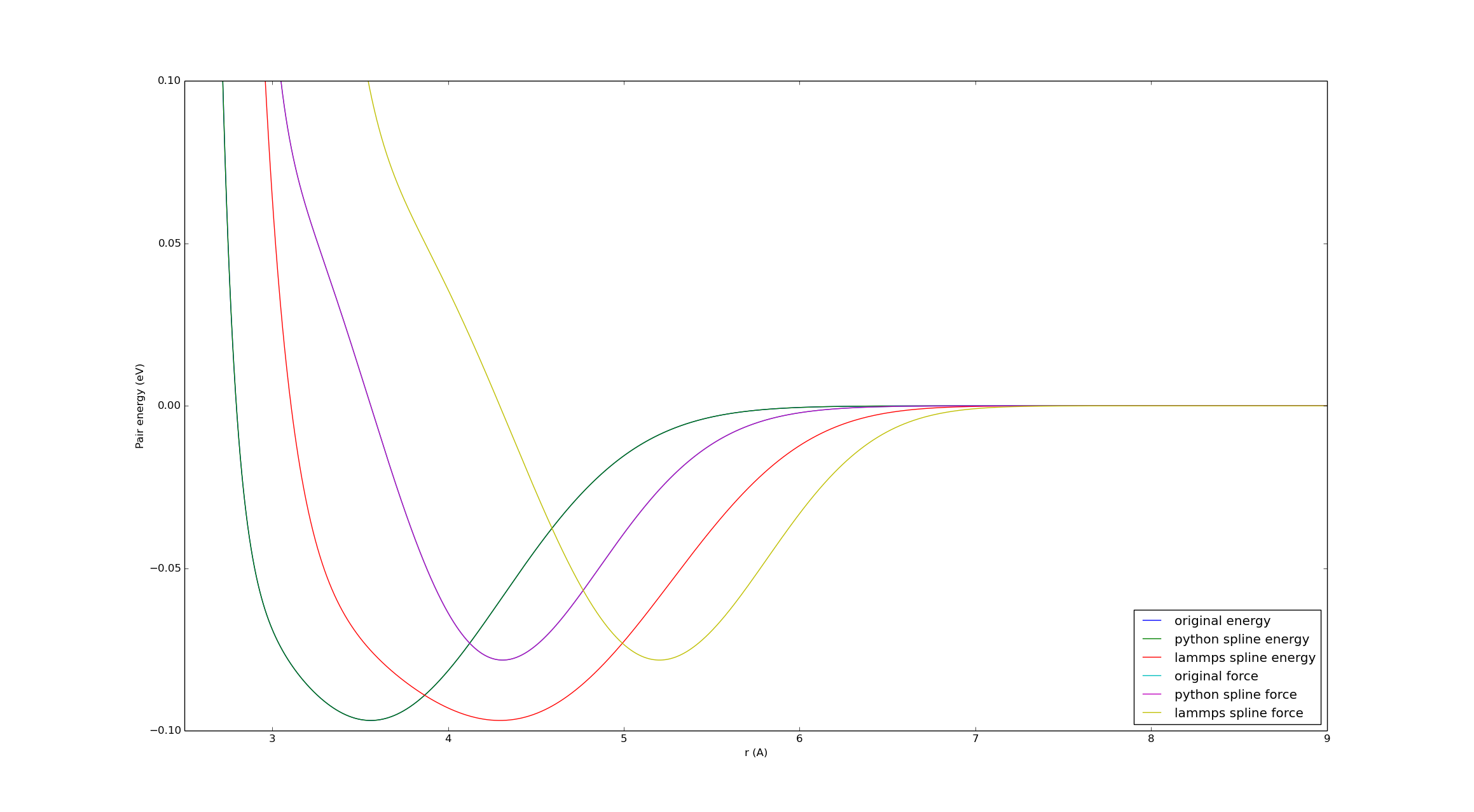
data.n2000 (268 KB)
in.tabulated (343 Bytes)
potential.table (273 KB)
I have changed the inner cutoff from 1 to 2.5 Angstroem and now it works
with the "RSQ" option!
All the needed LAMMPS script files with working parameters are attached.
The interpolation which does LAMMPS looks smooth, but unfortunately wrong.
no. LAMMPS is right, *your* input/table is wrong. the table has linear
spacing, thus must set the R option not RSQ.
axel
yep, thanks! now it works fine! I have noticed that if to do the tabulation of the potential in RSQ mode one needs lesser amount of discretization points (compared to the R mode) in order to achieve a good convergence!
Do I understand correctly that the performance of LAMMPS should not (or to a very little extent) depend on the number of discretization points?
yep, thanks! now it works fine! I have noticed that if to do the tabulation
of the potential in RSQ mode one needs lesser amount of discretization
points (compared to the R mode) in order to achieve a good convergence!
Do I understand correctly that the performance of LAMMPS should not (or to a
very little extent) depend on the number of discretization points?
that is a question that only some carefully crafted benchmarks can
answer with confidence.
axel.