Hi,
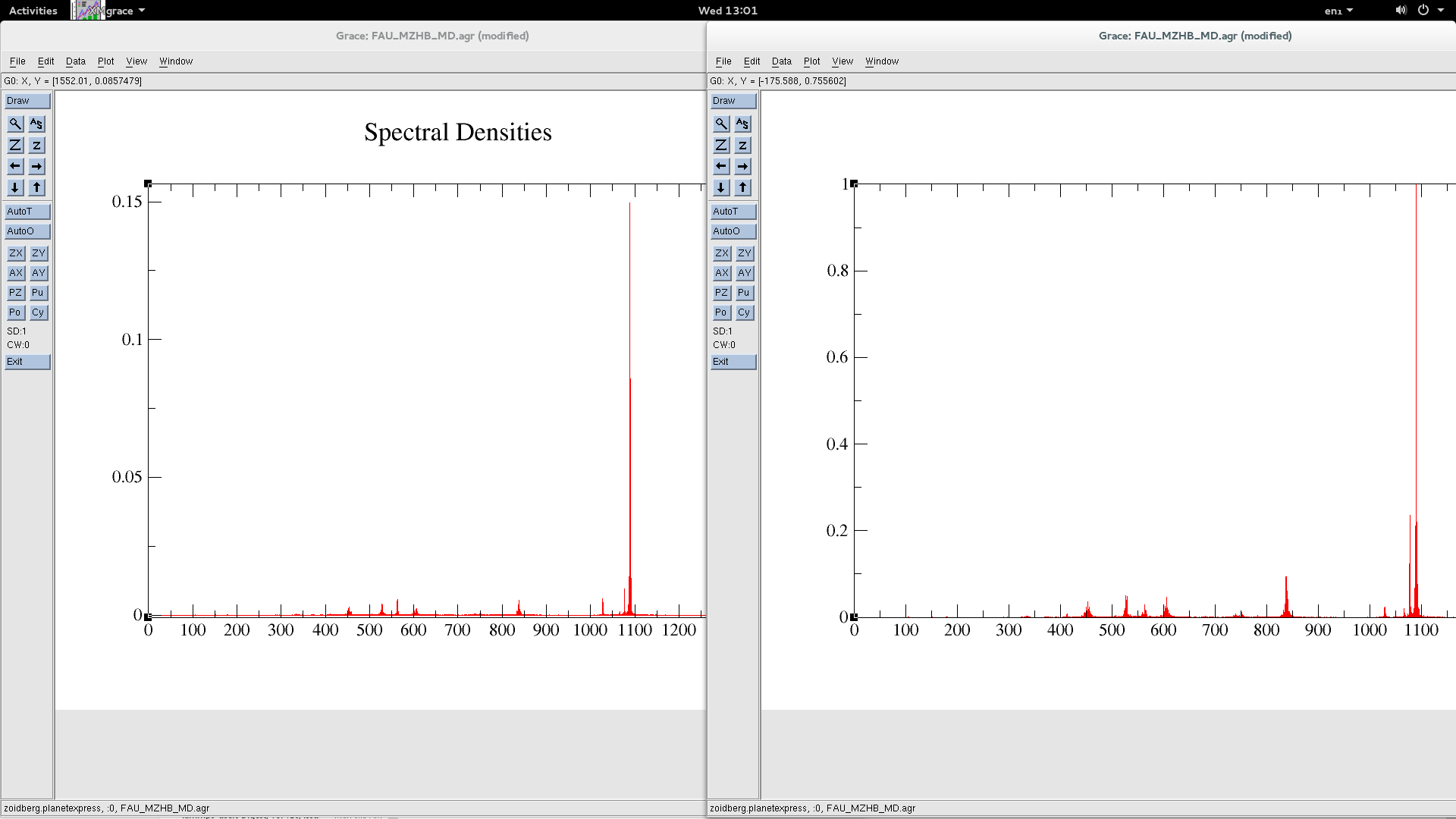
Thanks very much for the reply of Steve and Aidan. i did what Aidan suggested, compare the spectrum from a simple force field, and i have attached the resulting spectrum, picture 1,(left - lammps, right - GULP)
the IR wavenumbers in this simple case matches with each other, relative intensity has sight difference, which may come from statistic errors, we don’t care as much as the wavenumbers. And i check the energy calculated by individual potential terms. all the other terms works fine, the issue lies on the improper term, the energy of the improper term calculated in lammps is actually three times the value in GULP.
my initial lammps input file is dumped from GULP geometry optimization, all the bond terms and angle terms are well defined, but the improper terms are not, so i use moltemplate to define the improper terms. i don’t understand where does this energy difference comes from.
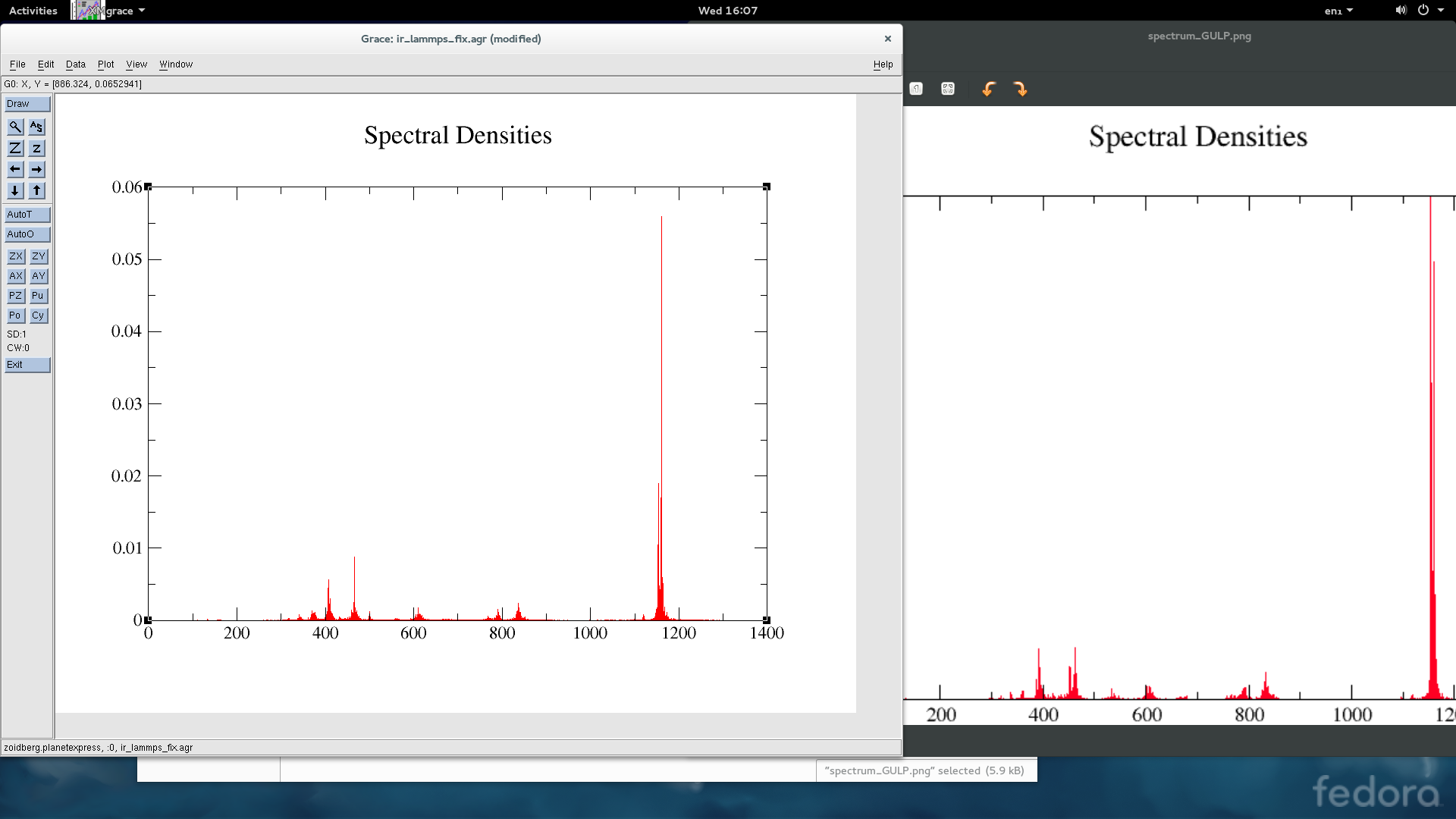
my currently solution is simply divide the improper coefficient by 3, that fix the energy difference. with this simple solution, i calculate the IR spectrum again, as is showed in picture2, (left - lammps, right - GULP) it improved a lot, but the peak around 400 still shift, and the peak at around 530 cm-1 appear only on the spectrum calculated with GULP.
any idea about what should check further? thanks for the help!!!
Best Regards
Jiasen Guo