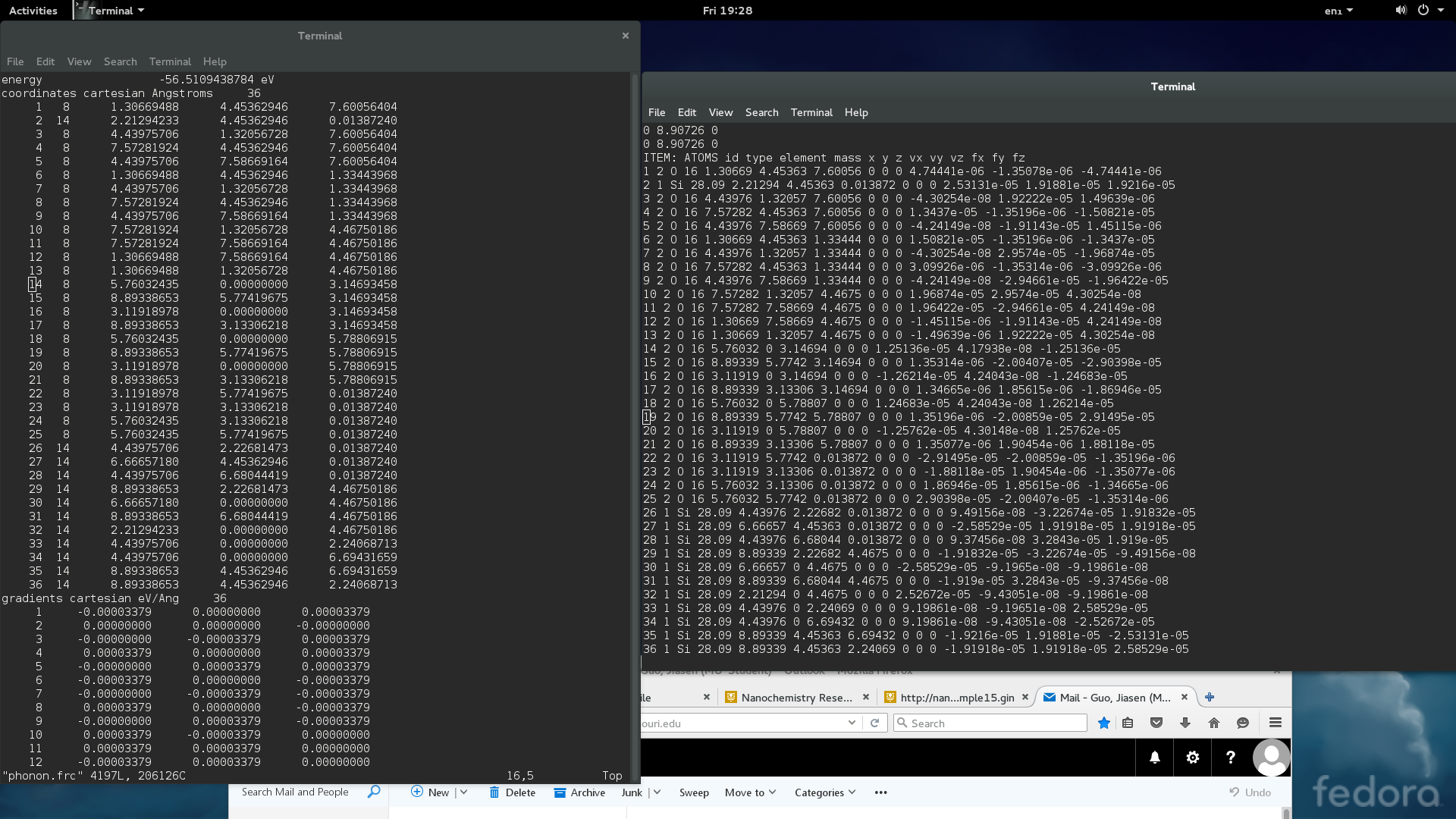
Hi,
thanks for the reply, Axel. i use a simple model (the model contains harmonic bond and angle term, couloum and l-j term), to test the consistency bewteen GULP and lammps, as showed in the picture 1. The atomic position are the same since it is dumped from GULP as final optimization geometry, but the per atom force are not. and when i check energy calculated by individual potential terms, it shows that the energy associated with bond term, angle term, and l-j term are matched between two codes, but the electrostatic energy differs about 0.05 ev, (which is the same situation when i work on the model that i really interested in ), i think the per atom force difference may come from the electrostatic term, but i have check the ewald summation accuracy and real space cutoff, both are the same with the parameters in GULP. any ideas about what should i check further? i attached my input script for lammps. thanks very much for your help!!!
And for that issue about improper energy which you pointed out “not scientific”, i realized that in my lammps data file, each SiO4 tetrahedral has 12 improper, 8 of which are redundant, so that the calculated improper energy becomes three time the real energy.( see below, the fourth column are Si atoms, each Si are included in 12 improper ) . i generate these impropers in moltemplate with the follwing command (with atom1 =Si, atom 2= O)
write_once(“Data Impropers By Type (class2_imp.py)”) {
@improper:type1 @atom:type1 @atom:type2 @atom:type2 @atom:type2 @bond:* @bond:* @bond:*
}
does anyone have a better way to define these class2_impropers?
Best Regards
Jiasen Guo
impropers

in.MZHB_SOD_minization (1.12 KB)