Dear Dr. Shan,
Thanks so much for your encouraging advice! For your last advices, I have the following uncertainties. If possible, please please please give me more advices. I really really really appreciate your help! Thanks very very much!
1 Your advices: Stopping at a step that is one step less than the multiples of fix reax/c/species’ Nfreq value is a typical error of fix reax/c/species. I already suggested you to try the Kokkos version.
I re-compiled lammps executable with Reax/c and kokkos packages.
However, when I got the following errors when I run reax/c/kk with the following settings:
“ERROR-1: Cannot yet use minimize with Kokkos (…/minimize.cpp:57)”
For this minimization error, I tried 2 separated simulation, 1st one for minimization only without kk, 2nd one for NVT equilibration with kk, however the ending potential energy of 1st case is different from the starting potential energy of 2nd case, can this situation be acceptable?
“ERROR-2: Increase MAXSPECBOND in reaxc_defs.h”
For this error, I am totally confused and stuck in here.
Setting in the input script:
pair_style reax/c/kk lmp_control safezone 4.0
pair_coeff * * ffield.reax Ni Cr Fe H O
fix QEQ all qeq/reax/kk 1 0.0 10.0 1.0e-6 reax/c
fix_modify QEQ energy yes
neighbor 2.5 bin
neigh_modify every 10 delay 0 check yes once no cluster no exclude none page 100000 one 2000 binsize 0.0
fix NVT1 all nvt temp 600 600 100 drag 0.2
fix SP all reax/c/species/kk 1 1 10 SCCspecies.out element Ni Cr Fe H O
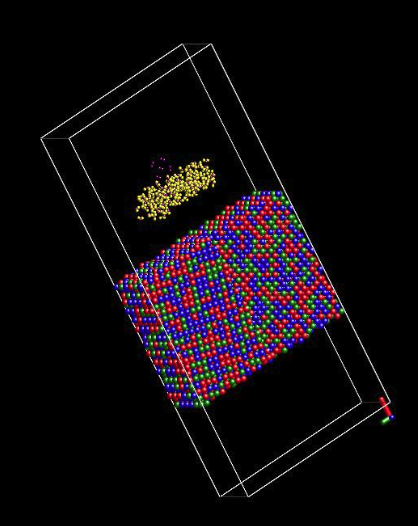
2 Your advices: Your alloy substrate is a block of atoms isolated in vacuum. Instead you should have a slab or surface geometry. Please consult with local experts if you need further assistance in this regard.
I re-built a bi-crystal Ni-Cr-Fe alloy as shown in the attached picture. How do you like this slab alloy model as shown in the attached picture?
This time, by the following commends in red, I only expend the box in x-direction, while the y and z surfaces of alloy were set to match the boundary of box by the following commends.
Setting in the input script:
lattice fcc 3.52
region box block 0 15 -10 10 0 6 side in
create_box 5 box
region left block INF INF -10 0 INF INF side in
lattice fcc 3.52 orient x 5 -9 0 orient y 9 5 0 orient z 0 0 1
create_atoms 3 region left
region right block INF INF 0 10 INF INF side in
lattice fcc 3.52 orient x 5 9 0 orient y -9 5 0 orient z 0 0 1
create_atoms 3 region right
read_data data1.watervmd add append offset 3 0 0 0 0 group medium shift 58 -15 0.5
read_data data4.oxygen add append offset 4 0 0 0 0 group medium shift 73 0 8.8
change_box all x scale 2 y scale 1 z scale 1
3 Your advices: How did you specifically “switch off the H-Ni, H-Cr, H-Fe, O-Ni, O-Cr, O-Fe bonds in the force field file by setting the corresponding bonding parameters to 0 0 0”? Which parameters did you change? Changing force field file is a tricky business – are you sure you have changed the correct parameters?
I did the changes only in the following part of the force field file. For the other parts, I changed nothing!
By the way, in the force field parameterization, there is bond information of Fe-Cr, Fe-Ni, but no bond information of Ni-Cr, for such background, can my Ni-Cr-Fe alloy cluster be still physical?
44 ! Nr of bonds; Edis1;LPpen;n.u.;pbe1;pbo5;13corr;pbo6 pbe2;pbo3;pbo4;n.u.;pbo1;pbo2;ovcorr
bonds
before
after