hello sir,
i am trying to simulate argon channel between two fixed gold plates, for my simulation. i am trying to use eam potential for gold for some reason the file is not recognized by the lammps.
i am posting the code here.
log data.85K_heating.log
units real
boundary p p p
atom_style atomic
region box block 0 150 0 6 0 6
create_box 2 box
region 1 block 0 1 0 6 0 6
region 2 block 149 150 0 6 0 6
region 3 block 2 4 0 4.7 0 4.7
region 4 block 147 149 0 4.7 0 4.7
Create Au atoms
lattice fcc 4.08
create_atoms 1 region 1
create_atoms 1 region 2
mass 1 196.966
pair_style eam
pair_coeff * * Au_u3.eam
Create ar atoms
lattice fcc 5.4
create_atoms 2 region 3
create_atoms 2 region 4
mass 2 39.948
pair_style lj/cut 8.5
pair_coeff * * .2381 3.41
velocity all create 85.0 1 dist gaussian
neighbor 2.5 bin
neigh_modify every 2 delay 10
fix 1 all npt temp 85.0 85.0 25.0 iso 20.0 20.0 100000.0
thermo_modify lost ignore flush yes
minimize 1.0e-4 1.0e-6 1000 10000
timestep 5
run_style verlet
thermo 500
dump 85Kheat all custom 5000 heating_sim_85K.dim.*lammpstrj x y z
thermo_style custom step pe press temp vol ke etotal density
thermo_modify lost ignore flush yes
run 100000
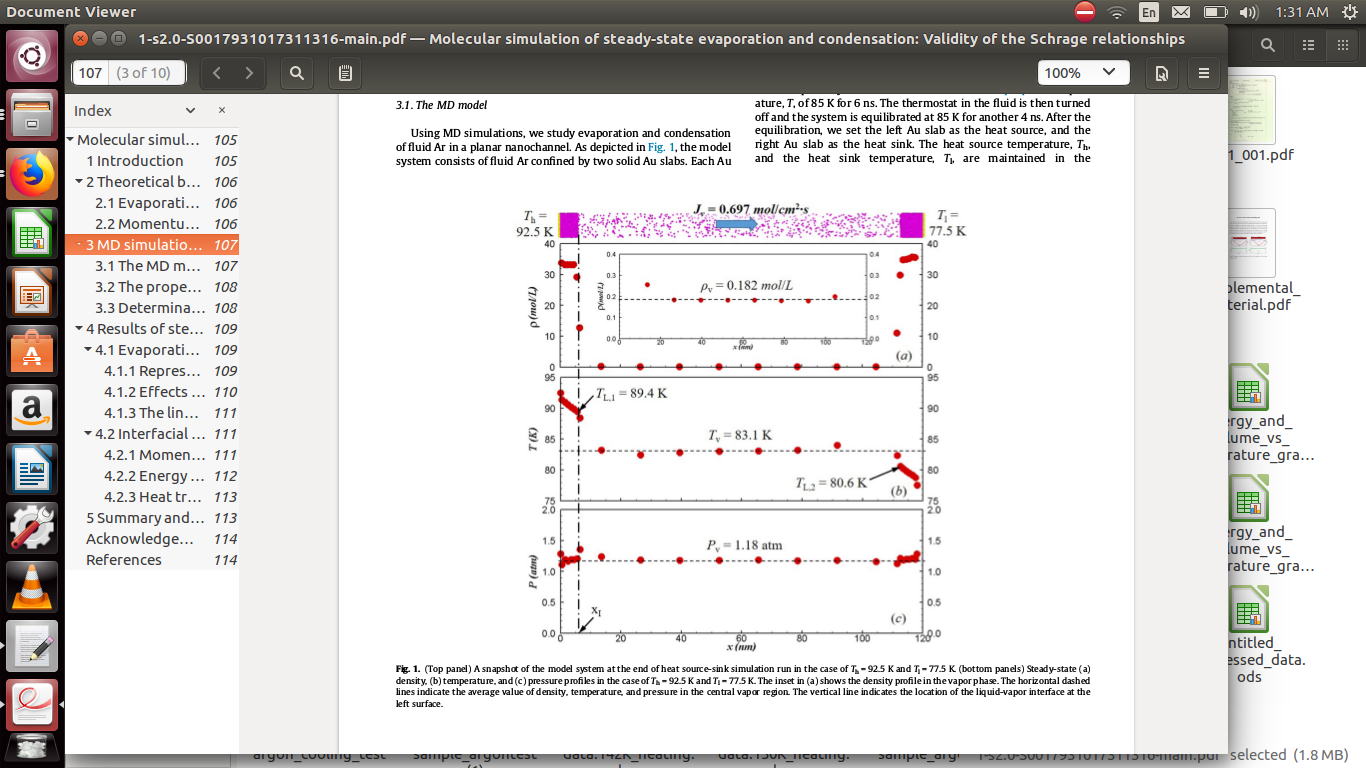
the above picture is the simulation result i am trying to replicate i need help.
thanking you,
nikhilesh jaladi
hello sir,
i am trying to simulate argon channel between two fixed gold plates, for
my simulation. i am trying to use eam potential for gold for some reason
the file is not recognized by the lammps.
that is because your second pair_style command will wipe out the effect of
the first (same for pair_coeff). if you want to have two pair styles
active at the same time, you have to use pair style hybrid.
there are lots of other, rather obvious issues, too:
- you are using "real" units, but most EAM potential files require using
"metal" units (including all of those bundled with LAMMPS)
- using a variable cell time integrator fix (i.e. fix npt) makes little
sense here, especially when coupled isotropically
- for a system like this, you must not ignore lost atoms. those are a sign
of a simulation gone bad. with all periodic boundaries, there should be no
atoms lost.
- the sketch in the image suggest, that you have non-periodic boundaries
in the direction of the gold slabs
- you need a way to immobilize those gold plates. they should be
thermalized differently, while the argon part should not be thermalized at
all
- your box definition doesn't look like it is commensurate with the gold
lattice spacing and thus you won't have proper continuation across periodic
boundaries
i strongly suggest to first figure out how to do a bulk argon system
simulation correctly (and in metal units!), i.e. setup, equilibration and
production with checking the results (e.g. g(r), self-diffusion) against
published data. then do the same with the gold setup, then only the gold
plates, and only if all of those were done successfully, i would try to set
up and run the combined system.
axel.