Dear Andrew,
I am trying to simulate a gold nanoparticle in lammps.
Okay.
(Disclaimer: I have no idea what potential you should use for this.)
I have generated a p1 cell of my molecule of interest and using the input
that is posted in this website
https://icme.hpc.msstate.edu/mediawiki/index.php/Uniaxial_Compression
however I can see the cell getting longer in the x direction but the
nanoparticle is not deforming.
You mentioned this web page:
https://icme.hpc.msstate.edu/mediawiki/index.php/Uniaxial_Compression
I don't think they are simulating a nanoparticle in this example.
I think they are simulating bulk aluminum. In that example, I think
the aluminum atoms completely fill the periodic boundary box. So when
they change the size of the box, the atoms move because they are
pushed on by the atoms in the neighboring cell due to periodic
boundary conditions.
I suspect your nanoparticle (a small crystal) does not fill the entire
boundary box. (It should not.) In that case, the atoms on the edge
of your small crystal will not interact with the atoms in the
neighboring cell. So when the box size and shape is changed, there is
no reason for the atoms to move. They don't feel each other.
If you want to deform a finite-size crystal, you can apply a force or
a strain to some of the atoms in that crystal using "fix move" OR "fix
addforce" :
http://lammps.sandia.gov/doc/fix_move.html
http://lammps.sandia.gov/doc/fix_addforce.html
In both cases, you would need to define a group of atoms at either end
of your nanoparticle and apply these fixes to those groups.
Especifically I want
to simulate the strain Vs stress curve but I have no luck into this.
Do you have a input for example that can be modified and used for my job?I have some questions regarding how to generate the inital coordinates.
msi2lammps is one option and using vmd.
If you simply want to create a finite-size rectangular fcc crystal,
there is nothing wrong with using LAMMPS script commands to build it,
as they did in the tutorial.
If you prefer to create a lammps DATA file, you could use either
VMD/topotools or moltemplate. I don't know the other tools well
enough to comment on them.
However I should say that unless you are trying to build a
nanoparticle with a strange shape, or mix it with other molecules,
there's no real reason to use a DATA file. The input script commands
in the tutorial above should work.
---- moltemplate ----
If, for some reason, you want to use moltemplate, then attached some
moltemplate files (.LT) files which can be used to generate the same
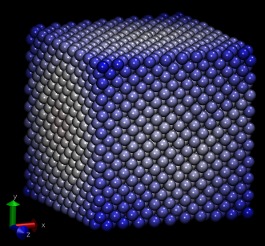
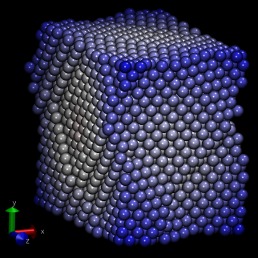
aluminum crystal used in Mark Tschopp's tutorial.
(Note: These posted files may seem unnecessarily complicated because
always prefer to use "atom_style full". I do this so that later on I
have the option to add molecules to the system, or apply charges on
the boundary of a surface. I assume this is what you plan to do. In
any case, I have tested these attached files and they appear to work
with lammps and moltemplate. The attached images were generated by
running lammps on these files. Note: I omitted the "Al99.eam.alloy"
file, because it is large, but you can find it using google.)
You can use these two ".LT" files to create a DATA file for an
aluminum crystal using these files using this command:
moltemplate.sh system.lt
This will generate three files: system.data, system.in.init,
system.in.settings.
Then you can run a lammps simulation using the input script "run.in"
(also attached).
lmp_ubuntu -i run.in
(The "run.in" file tells LAMMPS to read these three files.)
If you want to increase the size of the periodic boundary box, edit
the "system.lt" file, and change the "Data Boundary" section. (Or, if
you prefer, then edit the DATA file which moltemplate creates.)
Cheers
Andrew
(Credit: The script commands and force-field settings in these files
were stolen from Mark Tschopp's tutorial.)
system.lt (788 Bytes)
al_cell.lt (1.69 KB)
run.in (2.14 KB)