Dear Lammps users,
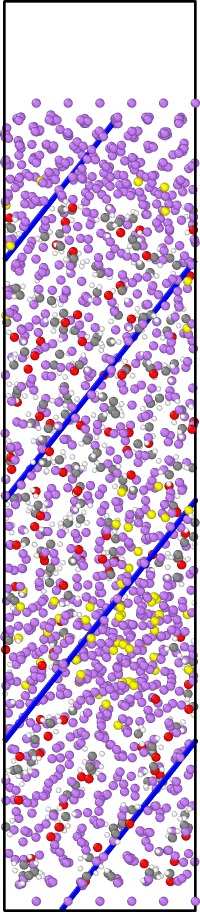
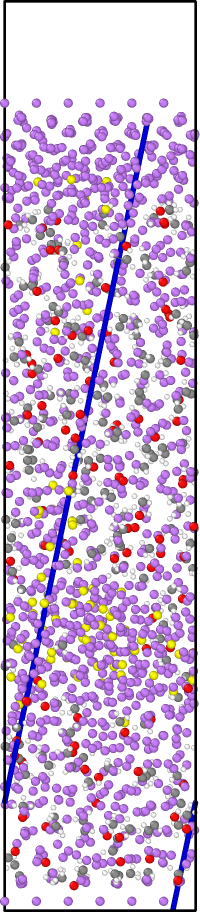
I am running a simulation of a system composed of two Li slabs (top and bottom) and an electrolyte mix between them. I am using the efield command to represent the effect of an electric field in the bottom slab (the idea is to try to see if there is any Li atoms transfer between the slabs because of the E field, when an amorphous SEI is forming close to one of the slabs), using the ReaxFF potential. The simulation runs well, but when I restart it (because of the reach of the time limit in the machine I am running) using the restart files generate by the restart command, there is an atom that just goes straight down in an almost perfect trajectory, even in the opposite direction of the efield applied (i.e. the atom doesn’t interact with any other atom just a shown in figures 1 and 2, like it is a totally inert atom or so) and kills the simulations because reaches the position of the fixed wall applied with the command fix wall/lj126 (the system has periodicity in X and Y directions, but doesn’t have in Z direction to avoid direct interaction between slabs).
For both the initial simulation and the restart one I am using one single node and requesting the use of 20 cores in the node.
The question is, is there any way to fix this problem? could it be related to the partitions of the task when running in one node/20cores? or could be related to any wrong specification in the input file?
I am attaching the initial and the restart input files, and a front and side view of the trajectory.
Thank you so much for your time and collaboration.
input.lammps (3.22 KB)
restart.lammps (3.24 KB)