Dear Dr. Axel Kohlmeyer,
Hello Dr. Kohlmeyer. Once again, I am deeply appreciative for your many suggestions and comments on LAMMPS during your busy schedule
I currently have a coarse-grained P3HT polymer chains where these chains form an aggregate that mimic a hairpin-like structure as shown in the below figure.
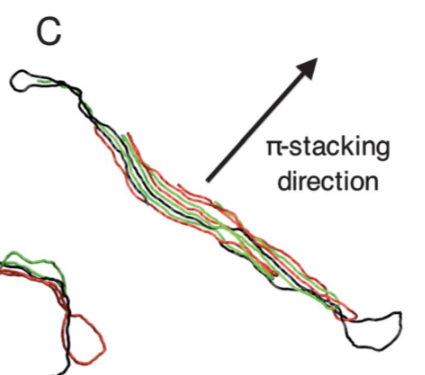
This aggregation is due to pi-pi interaction which is realized via LJ potential applied between Type 1 (backbone monomers) coarse grain beads that constitute the P3HT chains.
There are Type 2 and Type 3 beads that constitute the P3HT chains but the pi-stacking will cause Type 1 bead from one chain to stack on top of Type 1 bead from another chain.
I would like to obtain the radial distribution function of Type 1 bead only along the pi-stacking direction.
I understand that RDF command in LAMMPS will find the target bead where the radial distance will span in every direction (spherical coordinate).
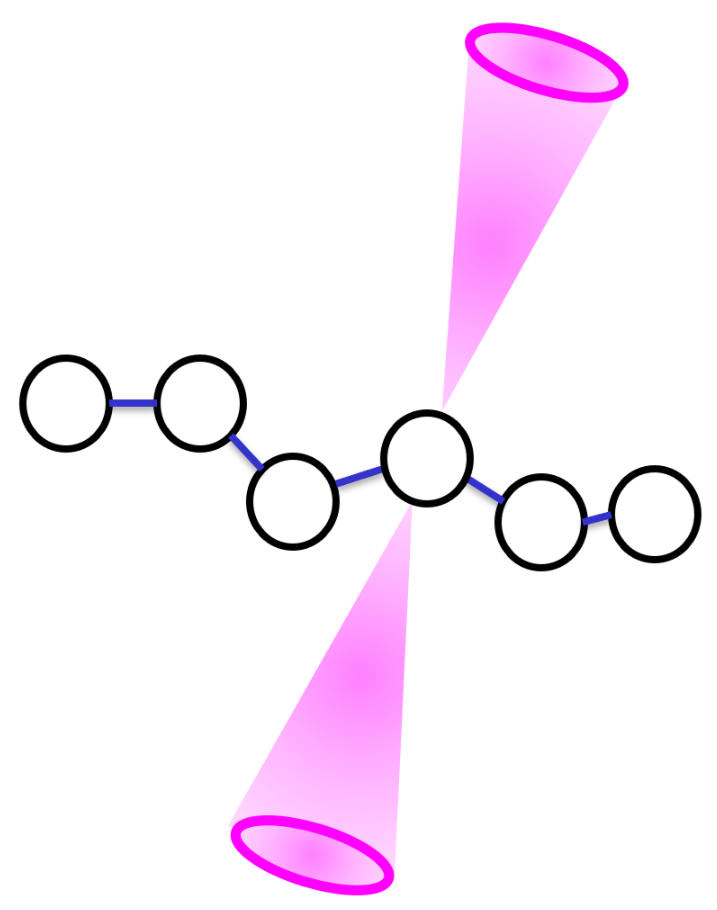
Is there a way in LAMMPS to restrict the finding P1 beads for RDF command such that the RDF command will only search for the desired Type 1 bead within the cone region as I have drawn in below figure. The cone region is always orthogonal to the connecting bonds that link the backbone monomers of the chain.
Once again, I would like to deeply thank you for your time in providing me with helpful many advices.
Sincerely,
Masato Koizumi