Dear all,
I am modeling the flexible water and flexible co2 placed in between two silica surfaces, with periodic boundary conditions at x-axis and y-axis and non-periodic boundary at z-axis (perpendicular to the silica surfaces). I am looking at the contact angles for the three phases system. The timestep was chosen as 0.25 fs. Both cuts off of LJ and Coulomb interactions are 12 Angstroms. The long-range Coulomb forces are calculated with pppm and with " kspace_modify slab 3.0 ".
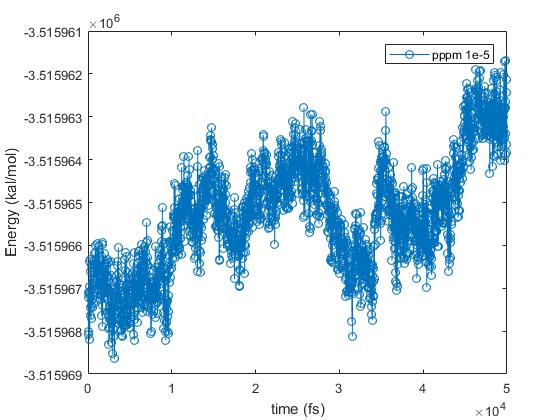
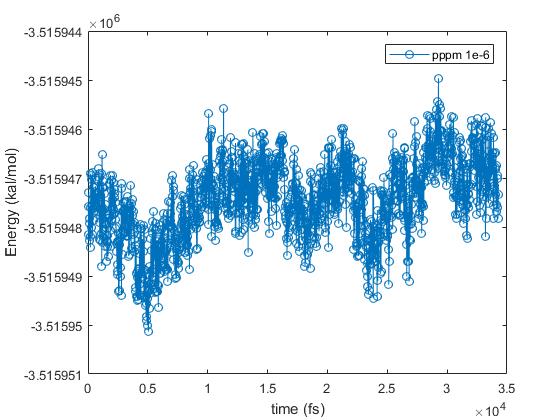
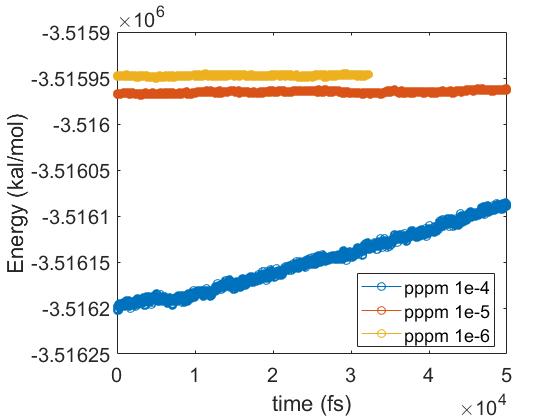
I have relaxed the system at 0.5 ns with Langevin and another 0.5 ns with nose-hoover thermostat with pppm 1e-6, with damping time of 50 fs. Then I have tested with different accuracy parameters of pppm in the NVE ensemble for 50 ps: 1e-4, 1e-5 and 1e-6. Using accuracy of 1e-5 is approximately 2 times faster than with 1e-6, but it gives a very small energy drift (see attached). I have calculated the ratio of root-mean-square fluctuations of energy to the averaged energy of the system, which is 3.8 e-7. I think this value is quite low. I have also attached individual plots of energy vs time for simulations with “pppm 1e-5” and “pppm 1e-6”, although the one with “pppm 1e-6” has not finished yet.
The whole simulations would need around 10 ns for measuring the contact angle, according to the literature. Using “pppm 1e-6” would need around 4 months for my system (around 100,000 atoms) at a computational node with 32 cores, although I have seen someone using an accuracy parameter of 1e-4 and with a larger timestep. Should I be worried about the small energy drift if I am choosing the accuracy parameter of 1e-5 and running the simulation under nose-hoover thermostats? If I have to stick with “pppm 1e-6”, are there any other methods to speed up the simulations? I have tried the r-RESPA for calculating the kspace at every 1fs which can speed up to around 2 times for “pppm 1e-6”. Varying the cut off of Coulomb interactions would not help much. I also want to calculate correct pressure so I did not try pppm/stagger.
Thank you for your time.
Regards,
Pengyu