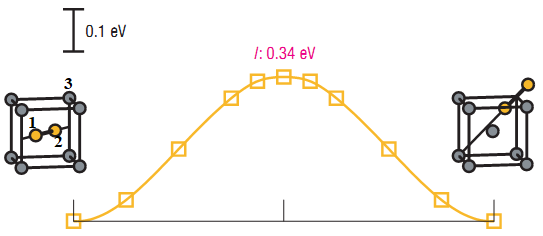
You don’t say how you are computing the energy changes or “forcing”
things to happen. This seems like a calculation for NEB, see the neb command
in LAMMPS. It can produce data for barrier plots like you show.
Yes, I am using NEB and i used the following commands for neb and non-neb atoms:
fix 2 nonneb setforce 0.0 0.0 0.0
fix 3 nebatoms neb 10.0
i don’t think that this is the problem and i do i saw the NEB command in LAMMPS.
(i know this is not a LAMMPS problem): I do not know what is the criteria in SIA migration.
i mean i do not know (according to the figure) that i should start with 10 atoms (one of them is an extra migrating atom) or with
9 atom (one of them is a migrating atom that is displaced from its position). However, my latter case gave me some better results!
maybe the crucial question is: Do we need to add an extra atom or not, in order to simulate migration energy using NEB?