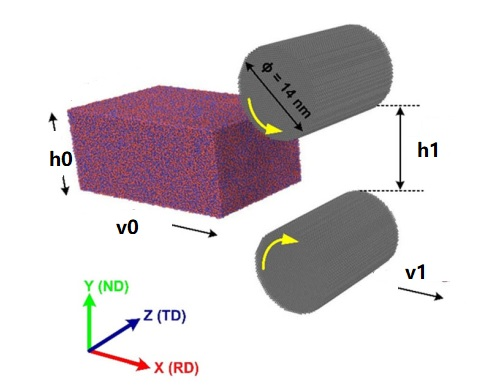
I am simulating the rolling process of nanocrystalline Al specimen using molecular dynamics simulations.When the cuboid workpiece passes through the cylindrical roll, the thickness direction(ND) becomes smaller, the length (RD) direction becomes longer, and the width direction remains the same. The workpiece speed v0 before entering the roll and the speed v1 after passing the roll are maintained as follows:
v1=v0*h0/h1
h0 is the initial thickness of the workpiece and h1 is the thickness after rolling
According to the speed relation, the speed of each atom in the direction of thickness should be different in the rolling process, and it will change with the change in the direction of thickness. How to set the speed of this process?
If you try to exactly set the velocity of the atoms then this will not be representative of a physical system, but a computer animation. You could instead apply a force in the RD direction that will move the group of atoms through the rollers. Where to apply the force (all atoms, front or rear sections, center of mass section) and when to stop applying the force (never, on first contact, when the workpiece is xx% of the way through) will be up to you.
When you output the result, you could correlate the speed and position of each atom in the RD direction to check if the results are what you expect, but you should not enforce the value of each atom’s velocity to be equal to the expected average.
Why not directly set up a non-equilibrium simulation of elongational deformation? fix deform command — LAMMPS documentation and so on. What you really want to know is the deformation behaviour of your workpiece – the means by which you impose it is immaterial, since your nanorollers can’t possibly be subatomically smooth perfect cylinders anyway.