Dear LAMMPS Experts,
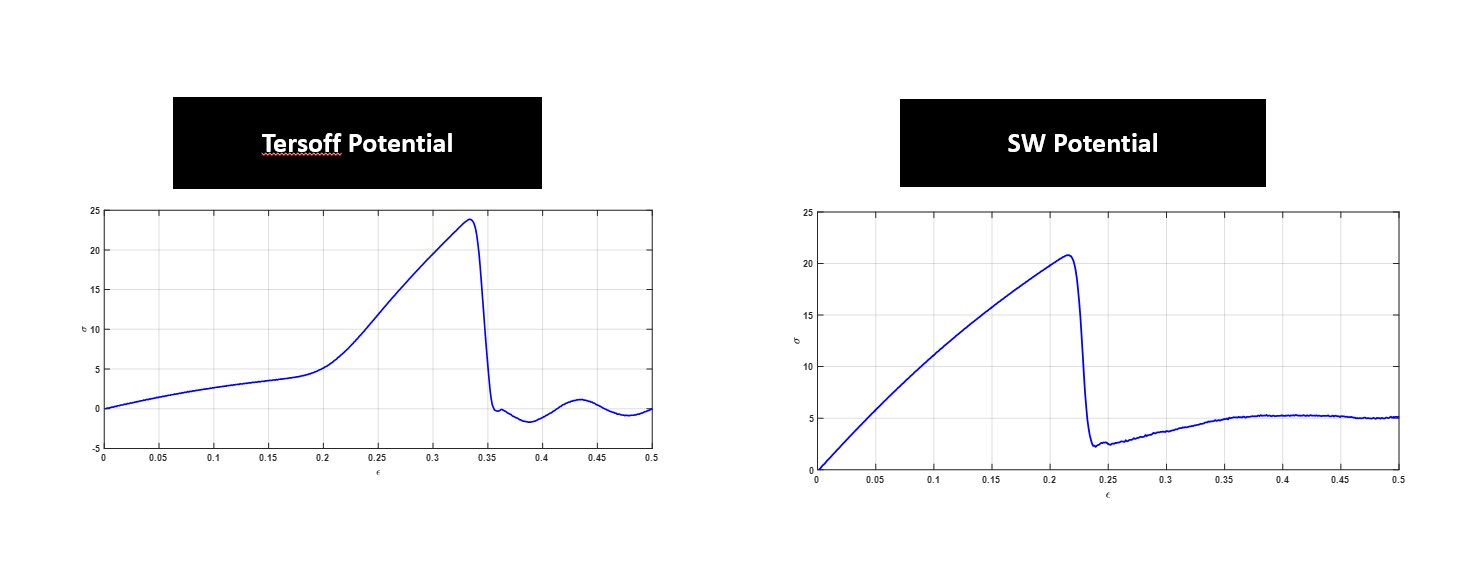
I am performing uniaxial tensile tests on Si(110) using the Fix Deform command. My concern in my results is the initial region of deformation using the Tersoff potential. Attached you can find a plot showing the stress-strain behavior of the Si sample under tension along it’s X direction and the only difference in my codes is the interatomic potential (SW vs. Terosff). As it’s obvious for the Tersoff plot, in the initial region (0–0.2 strain) compared to SW potential, stress does not increase which is sth different from the plots I have found in the literature. Does anybody know what might be the reason/explanation behind this behavior in my simulations?
I can share my code with you if needed.
Kind regards.
Hi,
Welcome to the wonderful world of non-equilibrium molecular dynamics. I am not as familiar with this specific case, but let me make a few general comments.
(1) Just because 2 different interatomic potentials (SW and Tersoff) have been parameterized (generally based on equilibrium properties) to “work” for a particular system does not guarantee that the potentials will agree with each other; or the real system for that matter.
(2) Make sure that your lammps data files are set up correctly: i.e. if you are deforming a system that is described with SW your system needs to be first equilibrated/annealed/thermalized with SW.
(3) In regards to your two specific plots, I see two significantly different graphs (across all strain) and would compare both to the real system to evaluate the applicability of your interatomic potential. A quick and easy check for you would be Young’s modulus.
In short, interatomic potentials don’t always agree with each other on all properties and they don’t always agree with the real system on all properties.
(These comments were all made assuming that your input script and input files are all in order. If you are finding that your SW results don’t agree with SW published results or your Tersoff results don’t agree with published Tersoff results that is a completely different story. We would need to see your input scripts and the published results to help with that problem.)
Best,
Dylan
Dear Dylan.
Thanks for the reply and comments, let me try to reply step by step as you suggested.
(1) I totally know how potential parameters work and indeed I do not expect them to be two plots on top of each other. There are people modeling uniaxial tension tests, for instance, this example (https://icme.hpc.msstate.edu/mediawiki/index.php/Uniaxial_Tension), so I was wondering if anybody has encountered sth similar in their simulations.
(2) I believe I have done proper energy minimization and pressure minimization and as I said the results are convincing for me in terms of finding the elastic modulus compared to the available E in the literature using SW interatomic potential. However, my only concern is by switching to Tersoff potential I am getting that different behavior in the initial region which I am curious to find an explanation for.
Attached you can find my sample code.
Best,
Sina
in.em (2.27 KB)