Hello everyone,
I am using LAMMPS window version 19 March 2020. I want just move one graphene layer to some distance. In the code, I have set velocity 0.5 A/fs and run for 40 steps with timestep of 0.25 fs. Then, at end, I should have displacement for 5 A.
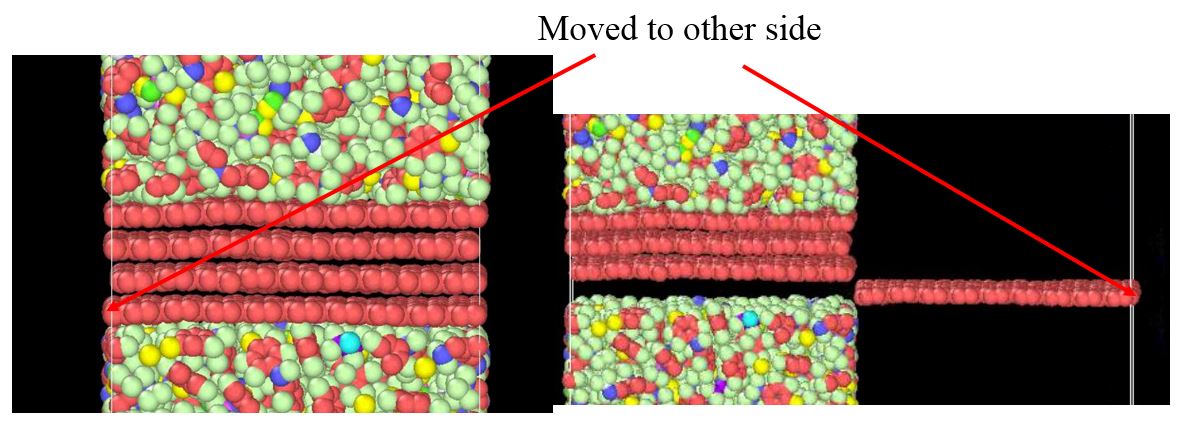
When I run the code, I found that for first step I am getting higher than 5 A, while from 2 to 40 steps, the graphene is moving at constant speed of 0.5 A/fs visualizing using Ovito 3.0 2020 version. Previously, I equilibrated the system using NPT ensemble where I was using periodic boundary conditions. As I want to pull the graphene. Here , I used boundary condition m f f. Below is code:
log “C:\one\log\justpull.txt”
echo screen
units real
dimension 3
boundary m p p
atom_style charge
read_data C:\one\datafile\data_1.txt
pair_style reax/c NULL safezone 2.0 mincap 200
pair_coeff * * …/potentials/ffield.Reax.latest C C O C O H C O C N H H
group Lowergraphene id 1:576:1
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
timestep 0.25
compute reax all pair reax/c
fix 1 all qeq/reax 1 0.0 10.0 1.0e-6 reax/c
variable eb equal c_reax[1]
variable ea equal c_reax[2]
variable elp equal c_reax[3]
variable emol equal c_reax[4]
variable ev equal c_reax[5]
variable epen equal c_reax[6]
variable ecoa equal c_reax[7]
variable ehb equal c_reax[8]
variable et equal c_reax[9]
variable eco equal c_reax[10]
variable ew equal c_reax[11]
variable ep equal c_reax[12]
variable efi equal c_reax[13]
variable eqeq equal c_reax[14]
fix 2 Lowergraphene move linear 0.5 NULL NULL units box
thermo 1
thermo_style custom step temp press pxx pyy pzz density enthalpy etotal pe ke epair evdwl ecoul v_eb v_ea v_elp v_emol v_ev v_epen v_ecoa v_ehb v_et v_eco v_ew v_ep v_efi v_eqeq lx ly lz
dump dumpdisp_1 all custom 1 C:\one\strjdisp\disp_1.lammpstrj id type x y z ix iy iz
run 40
write_data C:\one\datafileloop\data_2.txt
unfix 2
undump dumpdisp_1
print "code just pull is run successfully "
When I did visualize carefully, I found that In the image, atoms present in -X edge of box are coming to +X edge in 1 st step. How can I fix this issue?
Thanks and best regards,
Ankit