I am using LAMMPS version dated 2 Aug 2023 to equilibrate a domain of monocrystalline CsSnI3 to a certain temperature. The interatomic potential was obtained from https://doi.org/10.1021/acs.jpcc.2c05775 and it comprises of Lennard-Jones and Coulombic terms (parameters listed in attached figure). First, I utilized ‘box/relax’ along with ‘minimize’ to obtain a structurally relaxed domain (5000 atoms). Next, I utilized the following script to equilibrate the crystal at 5 K in the nvt ensemble.
timestep 0.2e-3 # in PICOSECONDS
neigh_modify every 1 delay 0 check yes
restart 4000 restart.perov
velocity all create 5 26745 mom yes rot yes dist gaussian
fix 1 all nvt temp 5 5 0.02
thermo 200
thermo_style custom step lz press temp
dump data all custom 200 dump.data id type mass x y z vx vy vz
run 20000
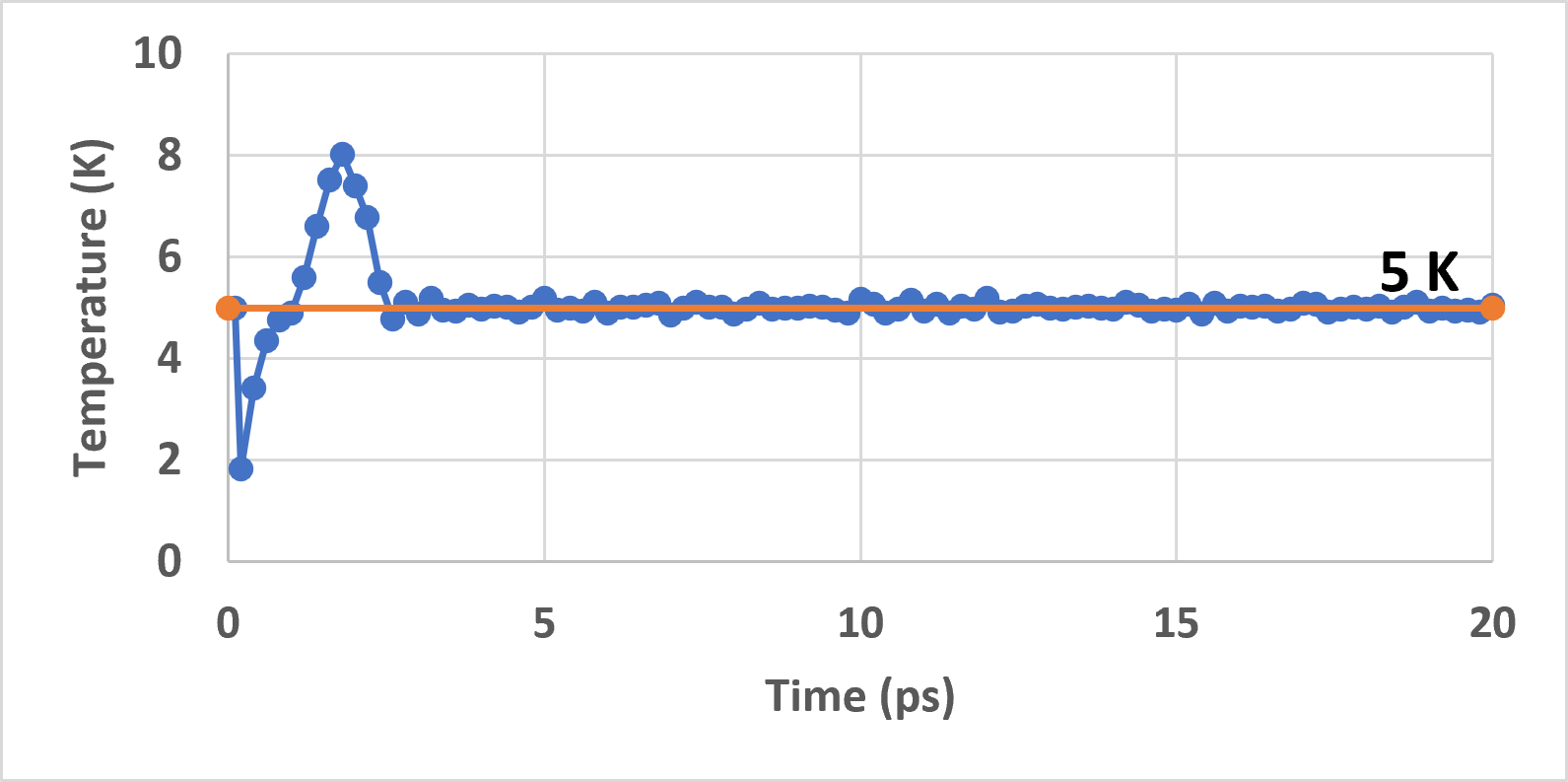
While conducting the nvt run, I noted that the temperature does not stay around 5 K and continuously increases upto a certain point and then drops at once to near zero (see attached figure). Any help in understanding this issue would be helpful.
I had also conducted a ‘fix-deform’ run to compare the total energy obtained based on this potential under volumetric compression to DFT results (also attached) which yielded good agreement. Thus, I am reasonably confident that the simulation parameters have been input correctly. However, the diverging temperature is puzzling.
To help people read your post, do not hesitate to have a look at the guidelines for posting. You’ll also find useful advices to help you track what’s wrong with your system.
For example:
are there any error in your log file? Can you please post it?
did you have a look at your simulation using any visualization software?
which units are you using? If you use the metal units, then a timestep of 2\cdot{}10^{-4} ps is very low for classical MD. Any reason for that?
why does you curve stops at 1ps? 20000\times{}2\cdot{}10^{-4} ps should be 4ps. Again, did your simulation crash?
why use the hybrid/overlay pair style? You can use the lj/cut/coul/long.
In which conditions did you perform a fix deform simulation? This is not informative without more context.
Hello @Germain, thank you for your time! Based on your suggestions, I utilized a timestep of 1e-3 ps instead of 2e-4 ps (it was a mistake) and utilized lj/cut/coul/long instead of the hybrid/overlay (I didn’t realize the former existed). Based on just these changes, I was able to equilibrate the domain in nvt at 5 K (see attached figure). The modified input script is as follows:
timestep 1e-3 # in PICOSECONDS
neigh_modify every 1 delay 0 check yes
restart 4000 restart.perov
velocity all create 5 26745 mom yes rot yes dist gaussian
fix 1 all nvt temp 5 5 0.1
thermo 200
thermo_style custom step lz press temp
dump data all custom 200 dump.data id type mass x y z vx vy vz
run 20000
Answers to some of your remaining questions are as follows:
are there any error in your log file? Can you please post it?
Yes, I have uploaded the log file for the run that used to give me the temperature divergence. Apart from the temperature divergence, the simulation solved to completion. log.perov.txt (8.6 KB)
did you have a look at your simulation using any visualization software?
Yes, but it did not help me decipher much.
In which conditions did you perform a fix deform simulation? This is not informative without more context.
I had conducted the fix-deform simulation to compare the total energy obtained using this potential under volumetric compression to DFT results. The script for this simulation is as follows:
neigh_modify every 1 delay 0 check yes
restart 50000 restart.perov
fix 1 all deform 1 x scale 0.875 y scale 0.875 z scale 0.875
thermo 200
dump 1 all xyz 1 dump.xyz
run 100000
As for why your settings were wrong I think that the use of hybrid/overlay was the most problematic part.
For the record, the manual explicitely states that:
Also for hybrid/overlay and hybrid/scaled mixing is only performed for pairs of atom types for which only a single pair style is assigned.
So your forcefield setup was bogus. It is recommended to provide all parameters explicitely with hybrid/overlay for a reason. In your case, hybrid/overlay was unnecessary.