Dear lammps users
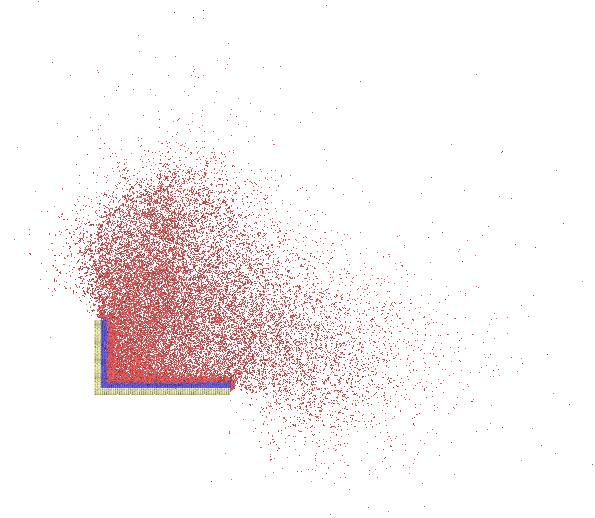
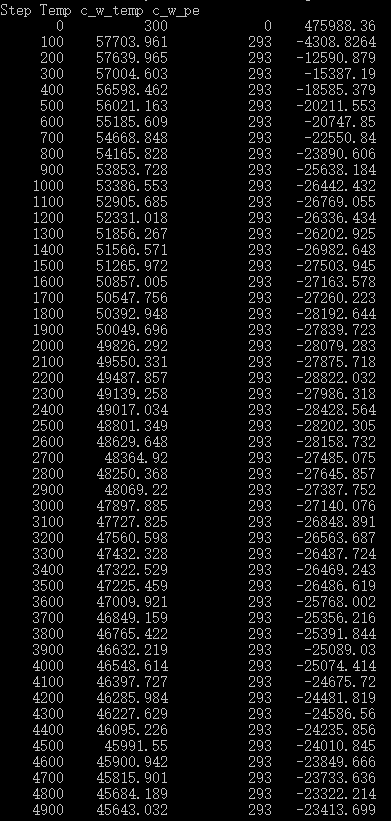
I built a monocrystalline silicon workpiece that needs to be balanced. However,the temperature of "work_newton"in the process of relaxation is particularly high and the atoms of the “work_newton” flew out ,I do not know what caused it .How can I reduce the temperature to normal stability ?Here is my Output data :
here is my program:
units metal
boundary s s p
atom_style atomic
region box block -400 0 -220 0 0 14
lattice diamond 5.431 orient x 1 -1 0 orient y 1 1 1 orient z -1 -1 2 #111 1-10
region work block -400 0 -220 0 0 14 units box
region work_newton block -360 0 -180 0 0 14 units box
region work_t1 block -380 -360 -180 0 0 14 units box
region work_t2 block -380 0 -200 -180 0 14 units box
region work_t union 2 work_t1 work_t2 units box
region work_b1 block -400 -380 -200 0 0 14 units box
region work_b2 block -400 0 -220 -200 0 14 units box
region work_b union 2 work_b1 work_b2 units box
create_box 3 box
create_atoms 3 region work
group work_newton region work_newton
group work_t region work_t
group work_b region work_b
group work region work
set group work_newton type 1
set group work_t type 2
set group work_b type 3
mass 1 28.0
mass 2 28.0
mass 3 28.0
pair_style tersoff
pair_coeff * * SiC.tersoff Si Si Si
neighbor 2.0 bin
neigh_modify delay 5
compute 2 work pe/atom
compute w_pe work reduce sum c_2
compute w_temp work_t temp
compute w_newton work_newton temp
fix b work_b setforce 0.0 0.0 0.0
velocity work_b set 0 0 0 units box
velocity work_newton create 300.0 5812775 temp w_newton
fix 1 work nve
fix res_w_temp work_t temp/rescale 10 293.0 293.0 10.0 1.0
fix_modify res_w_temp temp w_temp
timestep 0.001
thermo 100
thermo_style custom step temp c_w_temp c_w_pe
thermo_modify temp w_newton
run 5000
Here is formed graphics :
Best wishes
Liu Qiqi