Dear Sir,
The error is this:-
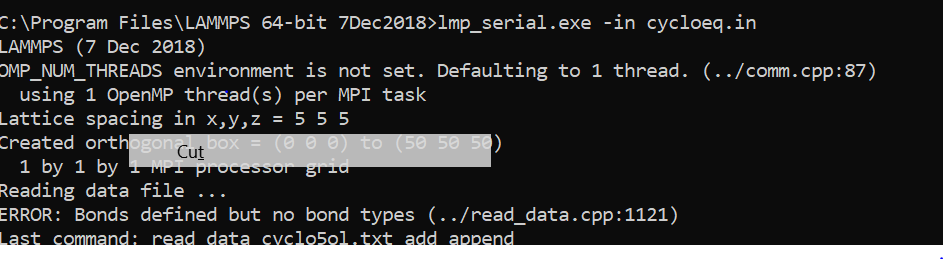
This is the complete file:-
#-----INITIALIZATION-----
units real
dimension 3
boundary p p p
atom_style full
#-----Atom definition-----
lattice sc 5
region whole block 0 10 0 10 0 10
create_box 1 whole
read_data cyclo5ol.txt add append
create_atom 0 box mol cyclopentanol
delete_atoms overlap “rc”
#-----Force Fields-----
pair_style lj/cut/coul/long 15.0 15.0
pair_modify tail yes
bond_style harmonic
angle_style harmonic
kspace_style pppm 1.0e-4
pair_coeff
1 0.066 3.5000000
2 0.066 3.5000000
3 0.066 3.5000000
4 0.066 3.5000000
5 0.066 3.5000000
6 0.170 3.1200000
7 0.030 2.5000000
8 0.030 2.5000000
9 0.030 2.5000000
10 0.030 2.5000000
11 0.030 2.5000000
12 0.030 2.5000000
13 0.030 2.5000000
14 0.030 2.5000000
15 0.030 2.5000000
16 0.000 0.0000000
bond_coeff
1 268.0000 1.5290
2 268.0000 1.5290
3 268.0000 1.5290
4 268.0000 1.5290
5 320.0000 1.4100
6 340.0000 1.0900
7 340.0000 1.0900
8 340.0000 1.0900
9 340.0000 1.0900
10 340.0000 1.0900
11 340.0000 1.0900
12 340.0000 1.0900
13 340.0000 1.0900
14 340.0000 1.0900
15 553.0000 0.9450
16 268.0000 1.5290
angle_coeff
1 58.350 112.700
2 58.350 112.700
3 58.350 112.700
4 50.000 109.500
5 37.500 110.700
6 37.500 110.700
7 37.500 110.700
8 37.500 110.700
9 37.500 110.700
10 37.500 110.700
11 37.500 110.700
12 37.500 110.700
13 37.500 110.700
14 55.000 108.500
15 50.000 109.500
16 37.500 110.700
17 37.500 110.700
18 33.000 107.800
19 33.000 107.800
20 37.500 110.700
21 37.500 110.700
22 58.350 112.700
23 33.000 107.800
24 37.500 110.700
25 37.500 110.700
26 35.000 109.500
27 37.500 110.700
28 37.500 110.700
29 33.000 107.800
30 58.350 112.700
31 37.500 110.700
neighbor 2.0 bin 464563 units box
neigh_modify every 1 delay 10 check yes
#-----Masses-----
mass 1 12.0107
mass 2 12.0107
mass 3 12.0107
mass 4 12.0107
mass 5 12.0107
mass 6 15.999
mass 7 1.008
mass 8 1.008
mass 9 1.008
mass 10 1.008
mass 11 1.008
mass 12 1.008
mass 13 1.008
mass 14 1.008
mass 15 1.008
mass 15 1.008
#-----Define setting-----
compute eng all pe/atom
compute eatoms all reduce sum c_eng
#-----Equilibriation-----
reset_timestep 0
group cyclopentanol type 1 2
fix mynpt cyclopentanol npt/asphere temp 300.0 300.0 100 tri 1.0 1.0 1000 mtk yes tloop 10.0 ploop 10.0
velocity all create 300.0 13430
run 0
velocity all scale 300.0
thermo 1
thermo_style custom step temp pe ke etotal press
min_style cg
minimize 1e-25 1e-25 2000000 2000000
variable PotentialEnergy equal epair
timestep 1.0
#-----Output-----
print “System is equilibiriated”
dump mf1 all molfile 100 cyclopentanol-.pdb pdb .:/Desktop
dump mf2 all molfile 100 cyclopentanol-.cfg cfg. :/Desktop
#-----RDF calculation-----
rerun cpmecyclopentanol-.cfg
special_bond coul 1e-50 1e-50 1e-50
compute 13 all rdf 150 cutoff 15
fix 07 all ave/time 100 10 1000 c_myRDF[] file tmp.rdf mode vector
print “The end is near . . .”
Best Regards,
Dhananjay