dear all
i recently do some researches on the peeling cnt from graphene
when the small system which contains about 2000 atoms was taken into computing
the result is nothing wrong
but when a large system which contains about 20000 atoms ,the result was wrong.
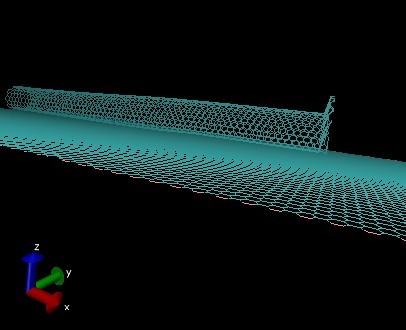
the pull group seperate from the whole cnt group at the beganing it was very different from the previous work
in addition the input file are similar
the input files and the image of the problem are in the afflix
could you find some problems from these files?
longing for your answer
yours xiaolin
I doubt anyone will deduce correct vs incorrect physics
from looking at your input files - it's up to you to insure
your simulation is doing the correct thing.
Suggestion:
Have you tried using the "move" command instead? This command pulls
some of atoms away slowly instead of all at once. If you pull slowly
enough, the break should not occur.