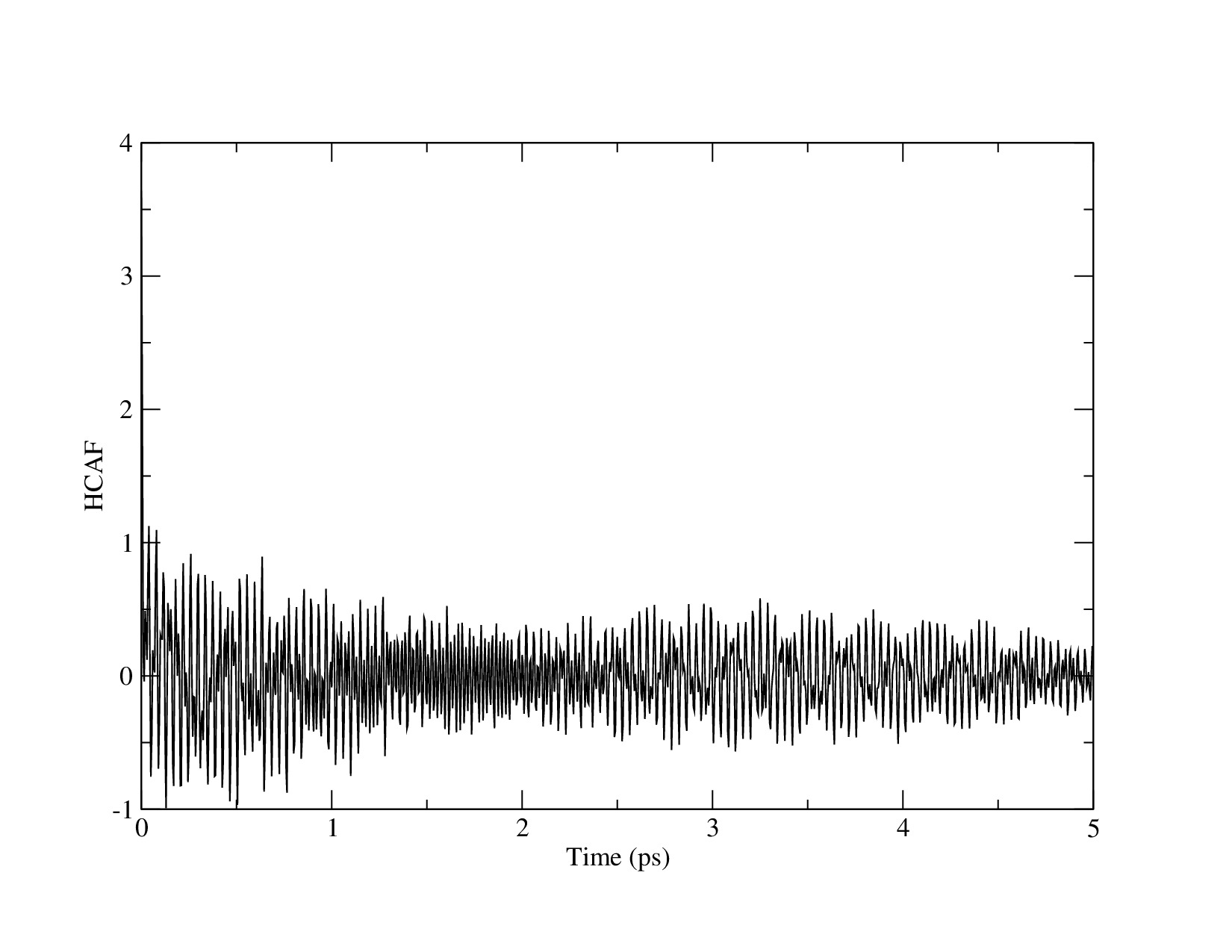
Dear lammps users,
Hello! I’m working on thermal conductivity prediction of Single Wall Carbon Nanotube by Charmm forcefiled. I equilibrated the CNT systems at first, and then did the thermal conductivity prediction by Green-Kubo method. However, I got a really small thermal conductivity of CNT. The thermal conductivity predicted result is only 0.6[W/mK]. I pasted my scripted at the end . In the thermal conductivity calculation. I tried different correlation length. However , it gave similar result about 0.6~10[W/mK]. I also tried to used Tersoff forcefield to do thermal conductivity calculation. But the result is still similar. I’m trying to find the problem in my scripts. Can anyone give me some comments?
So much thanks for the help.
Kerwin
Thermal conductivity calculation script in charmm forcefield:
variable T equal 273
variable dt equal 0.5
variable V equal vol
variable p equal 1000 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
#convert from Lammps real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal {kCal2J}*{kCal2J}/{fs2s}/{A2m}
setup problem
units real
dimension 3
atom_style full
neighbor 2.0 bin
neigh_modify every 2 delay 0 check yes page 100000
pair_style lj/charmm/coul/long 10.00 10.10
kspace_style pppm 1.0e-6
bond_style harmonic
angle_style charmm
dihedral_style charmm
improper_style harmonic
kspace_modify gewald 0.225
atom_modify sort 0 0.0
read_restart heatflux-2-run01
velocity all scale $T
thermal conductivity calculation
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom NULL virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix 1 all nve
fix JJ all ave/correlate $s $p d &
c_flux[1] c_flux[2] c_flux[3] type auto &
file profile.heatflux-run02 ave running
dump 2 all xyz 1000 xyz-2000.file-run02
dump 3 all custom 1000 dump_vmd-thermal-run02.lammpstrj id mol type x y z ix iy iz
variable scale equal {convert}/${kB}/$T/$T/$V*s*{dt}
variable k11 equal trap(f_JJ[3]){scale}
variable k22 equal trap(f_JJ[4])*{scale}
variable k33 equal trap(f_JJ[5])${scale}
thermo $d
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33 density press etotal
timestep 0.5
restart $d heatflux-1-run02 heatflux-2-run02
run 5000000
variable k equal (v_k11+v_k22+v_k33)/3.0
variable ndens equal count(all)/vol
print “average conductivity: $k[W/mK] @ T K, {ndens} /A^3”
reset_timestep 0
Thermal conductivity calculation script by tersoff forcefield:
Variables
variable input index lammps_data01
INITIALIZATION
units metal
boundary p p p
atom_style atomic
variable T equal 300.0
variable dt equal 0.0002 # ps
variable p equal 1000 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
thermo 100000
timestep ${dt} # ps
ATOM DEFINITION
read_data ${input}
FORCE FIELD
pair_style tersoff
pair_coeff * * /home/tntech.edu/czhan42/lammps-16Mar18/potentials/SiC.tersoff C
SETTINGS
neighbor 2.0 bin
neigh_modify delay 1
Calculating Volume
variable diameter equal (sqrt(3)1.42sqrt(6^2+6*6+6^2))/PI #(6,6) armchair CNT; assuming C-C bond distance of 1.42
variable radius equal {diameter}/2
variable height equal "lz" #Height is equal to z-dimension length
variable V equal 2*PI*{radius}3.4${height}
Convert from metal units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable eV2J equal 1.602176565e-19
variable A2m equal 1.0e-10
variable ps2s equal 1.0e-12
variable convert equal {eV2J}*{eV2J}/{ps2s}/{A2m}
group Carbon type 1
------------------------------------------------------------------------------
variable temp equal temp
output for VMD
dump 1 all custom 10000 dump.uhmwpe-coord id type x y z
dump 2 all xyz 10000 dump.uhmwpe-xyz-format
dump_modify 2 element C
— Relaxation —
fix 51 all nvt temp $T $T 0.1
run 100000
— Thermal Conductivity Calculation —
unfix 51
fix 51 all nve
thermo d
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom NULL virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/$V
variable Jy equal c_flux[2]/$V
variable Jz equal c_flux[3]/$V
fix JJ all ave/correlate $s $p d &
c_flux[1] c_flux[2] c_flux[3] type auto file profile.dat ave running
variable scale equal {convert}/${kB}/$T/$T/$V*s*{dt}
variable k11 equal trap(f_JJ[3]){scale}
variable k22 equal trap(f_JJ[4])*{scale}
variable k33 equal trap(f_JJ[5])${scale}
thermo_style custom step temp cpu v_Jx v_Jy v_Jz v_k11 v_k22 v_k33 density press etotal
run 5000000
variable k equal (v_k11+v_k22+v_k33)/3.0
print “average conductivity: $k[W/mK] @ $T K”