I would like to run a simuation of tip5p along with a rigid force field like epm2. I can see in the documentation that there is a suitable pair_style and kspace_style for the tip4p and I wonder if this is going to work with the epm2 or I should use a lj/cut/coul/long style instead.
I would like to run a simuation of tip5p along with a rigid force field like
epm2. I can see in the documentation that there is a suitable pair_style
and kspace_style for the tip4p and I wonder if this is going to work with
the epm2 or I should use a lj/cut/coul/long style instead.
tip4p is tip4p. it doesn't care about the rest. the tip4p pair style
operates as lj/cut/coul/long for all atoms that are not labeled as
tip4p oxygen/hydrogen.
it certainly, is also possible to just use lj/cut/coul/long for, but
then in combination with fix rigid(/small) instead of fix shake.
the major reason to have a special tip4p variant is that it can
implemented with 3 particles per water instead of 4 by shifting the
oxygen position back and forth and projecting the resulting forces on
the hydrogens. this can result in a significant speed increase.
Thanks for the quick answer. When simulating, let's say spc with epm2, I am using the exact case you mention: lj/cut/coul/long and fix rigid. Of course I am going to have some test runs with any variation but I just felt it was a nice thing to be mentioned and stored in the lammps mail data base.
Thanks again,
Otto
Actually you cannot use lj/cut/coul/long because as I can see, mass must be greater equal to zero. So the only way is the lj/cut/tip4p/long that will treat the waters as described but the other molecules as lj/cut/coul.
Otto
Actually you cannot use lj/cut/coul/long because as I can see, mass must be greater equal to zero.
So the only way is the lj/cut/tip4p/long that will treat the waters as described but the other molecules as lj/cut/coul.
Axel, that's the way I had it running too. Of course such a difference in mass is not going to make a difference but I think the most sophisticated way of implementing this is by the tip4p pair. Nice.
But after some thousand streps lammps crushes with the error: TIP4P hydrogen is missing. The most strange thing is that it isn't crashing always after the same number of steps.
To add here that I am using fix rigid/molecule to keep both water and CO2 rigid and I only use the bond style commands because It doesn't run without tem.
I tried to find the problem by looking at the source code in the appropriate routine but I still cannot see why it crushes. Do you have any ideas?
Thanks a lot, as always
But after some thousand streps lammps crushes with the error: TIP4P hydrogen is missing. The most strange thing is that it isn't crashing always after the same number of steps.
To add here that I am using fix rigid/molecule to keep both water and CO2 rigid and I only use the bond style commands because It doesn't run without tem.
I tried to find the problem by looking at the source code in the appropriate routine but I still cannot see why it crushes. Do you have any ideas?
there is too little information here.
what is your time step? and related to that, what is the behavior of
your potential energy around the time when you run into trouble?
what are your neighbor list update settings?
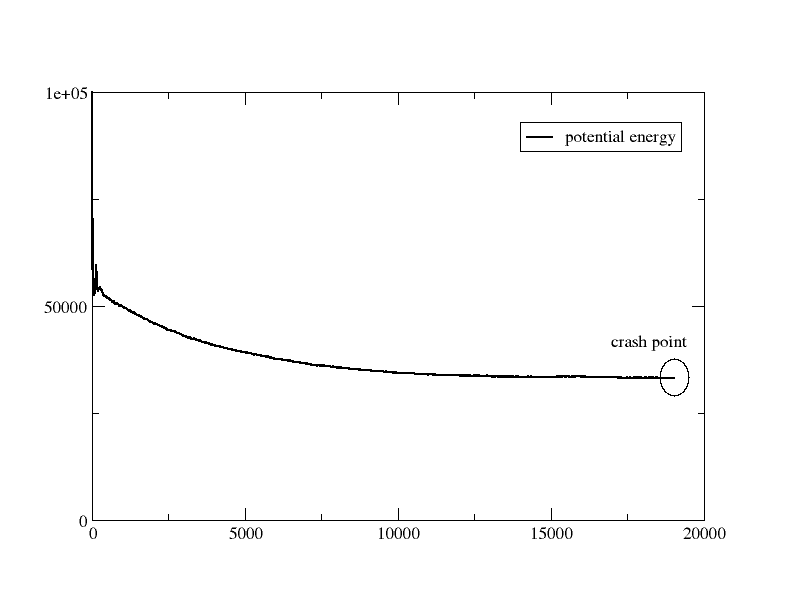
Thanks for the quick answer. Find attached a draft plot of the pe vs simulation time. I am performing test with various timesteps. I start with small ones (0.05) and going progressively to 0.5. I think that the problem is in my initial conformation. Maybe is too tight or something.
Thanks for the quick answer. Find attached a draft plot of the pe vs simulation time. I am performing test with various timesteps. I start with small ones (0.05) and going progressively to 0.5. I think that the problem is in my initial conformation. Maybe is too tight or something.
it doesn't look like it. if that was the case your energy would go up,
but it shows typical relaxation behavior.
this graph makes the neighbor list settings more of a problem. you can
be "missing a hydrogen", if your neighbor list skin is too small
and/or you update the neighbor lists too infrequent.
a) do a few run commands in your script each only for 5000 steps and
have a close look at the neighbor list statistics at the end,
particularly the Dangerous builds. it should be 0, ideally.
b) change to: neighbor 2.0 bin
and see if it makes a difference.
Axel,
Having set the neighbor to 2.0 bin doesn't seem to solve the problem. I can see my simulation crashing just a few steps later.
Also, my other test run, with the tip4p2005 model implemented as a rigid 4atom body (mass of M site=0.0000000001 and charged instead of O) seems to severely overestimate the density of the water.
I saw in your example link that we have pretty much the same data and in files. Have you seen a "correct" density with this way?
Axel,
Having set the neighbor to 2.0 bin doesn't seem to solve the problem. I can see my simulation crashing just a few steps later.
since i don't have a crystal ball and can't read minds, i cannot tell
you why. there obviously is something wrong, but you are the only
person that has the information what is going on. your vague
descriptions (it crashes) are not very helpful.
Also, my other test run, with the tip4p2005 model implemented as a rigid 4atom body (mass of M site=0.0000000001 and charged instead of O) seems to severely overestimate the density of the water.
overestimate with respect to what?
I saw in your example link that we have pretty much the same data and in files. Have you seen a "correct" density with this way?
the input you saw is part of my list of regression test library. their
purpose is not to produce meaningful science, but to make sure that
LAMMPS produces consistent results under different conditions
(different compilers, flags, platforms, parallelization, number of MPI
vs. OpenMP tasks etc.) and helps to spot bugs in the /omp or /opt
versions of the pair styles. it assumes that the original
implementation was correct when the original test was done (unless
proven wrong, of course)
It crashes with the same error of TIP4P missing Hydrogen.
As far as the other runs, they overestimate the density for the specific conditions according to the original paper by J. L. F. Abascal and C. Vega at J. Chem. Phys. 123, 234505 (2005). I can see that this is a sample library and I agree that you way follows the common sense (I did it that way myself).
My basic problem is that I cannot understand the "missing hydrogen".
It crashes with the same error of TIP4P missing Hydrogen.
As far as the other runs, they overestimate the density for the specific conditions according to the original paper by J. L. F. Abascal and C. Vega at J. Chem. Phys. 123, 234505 (2005). I can see that this is a sample library and I agree that you way follows the common sense (I did it that way myself).
My basic problem is that I cannot understand the "missing hydrogen".
you get the "missing hydrogen" error, when you have an atom that is
owned by the domain you are in interacting with a water molecule owned
by a neighboring domain (a ghost). those coordinates get updated and
communicated only with a given cutoff, which depends on the real
cutoff and the neighbor list skin. now if the oxygen is communicated
but one of the hydrogens not, you get this error. that is usually an
indication of neighborlists not being updated properly or some bad
physics (atoms getting too close) happening that extremely accelerates
a molecule, so that is passes through the communication stencil.
Please try the following piece of input file for TIP4P/2005 rigid. It has been verified (by me) that it produced consistent data with Vega’s group and NIST data.
I did not follow the whole conversation, but just a piece of advice, always start something very simple, then you proceed. If I were you, I will try to reproduce bulk water properties before going to add something else into the system.