I plan to conduct uniaxial tensile simulation of a polymer-solids system in z-direction, as shown in the following literature picture:
The idea is to fix the top region polymer layer, pull it away from the solids surface, equilibrate and measure the stress before the polymer is displaced again to plot stress-strain curve. (I’m not sure why the bottom solids region need to be fixed rigid too), May I know what is the command in LAMMPS to make it work? I try the fix deform command before but it doesn’t work. Only box boundary will move but not the polymer layers.
I check from the following thread using change_box command:
However, change_box command mentioned specifically changing the box boundary. Is the change_box command applicable to pulling atom too?
It is difficult to make specific comment on such a vague description and without seeing your input. But your use of the term “rigid” is suspicious. Most likely, you are confusing “rigid” with “immobile” (like many people do, including the authors of publications) and you are using fix rigid where you should not, but rather define groups of atoms based on regions and only apply time integration to the atoms outside the immobile region(s).
The change_box command can do all kinds of manipulations of a box including the box dimensions and tilt. Details are (obviously) in the LAMMPS manual. The main difference between fix deform and change_box is that the former operates gradually during a simulation run while the latter is applied instantly and can only be used in between runs.
Unfortunately, the author never mention the LAMMPS code he is using and I do not receive reply from him. I believe he refers the term “rigid” but should be using the setforce to make the group of atom immobile.
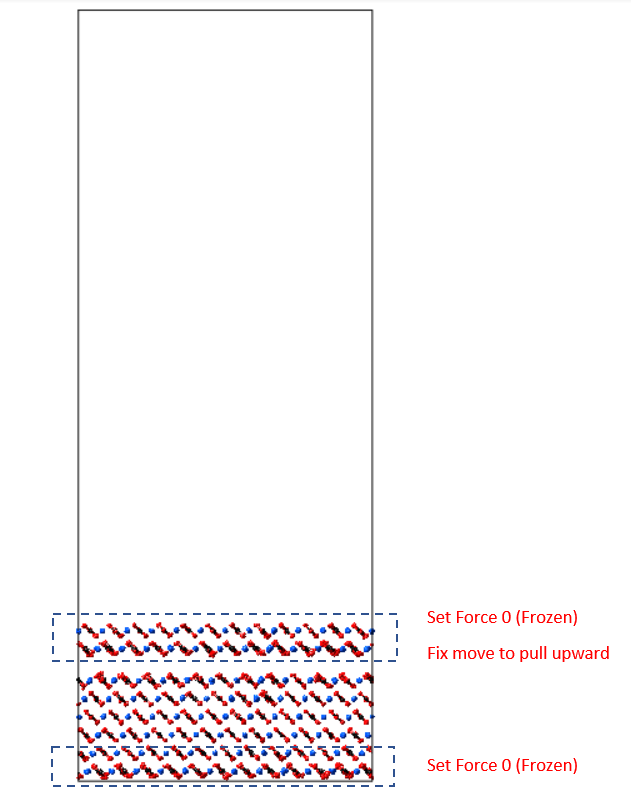
Below is my some input script when running the deformation simulation. I have declared the top polymer region and bottom solids region as fixed group to make sure they are immobile. The remaining movable atoms as movable group:
fix 1 movable npt temp 300 300 1 x 0 0 1 y 0 0 1 drag 1
fix 2 fixed setforce 0.0 0.0 0.0
fix 3 all deform 100 z erate 0.00001 units box remap x
run 100000
I will expect some output as the one from literature:
However, only the box will deform but the polymer will not move at all. The only way this command work is, when I do not freeze any atoms, and let the polymer layer equilibrates freely across PBC in z-direction before the deformation run. I will prefer to have vacuum region like in the literature, and only pull the polymer layer away gradually to measure the tensile stress…
After reading the fix move command, I think this is the command I’m looking for… Thanks!
One more question is, if we see from the fix deform command: fix 3 all deform 100 z erate 0.00001 units box remap x
we can select the timestep for the system to deform, like deform at every 100th timestep. I wonder if using the fix move command: fix 1 top_polymer move linear NULL NULL 1.0 units box
how can I do the displacement of the atoms at certain timestep interval only? It seems like with this velocity, the atoms will be displaced 1A every timestep…
There really is no need for that, you just reduce the velocity accordingly.
Otherwise you will have to do many short runs in a loop and execute displace_atoms in between.
I personally always prefer continuous motion over sudden displacements. The latter can induce undesired phonons.
fix tensile subset_top move linear NULL NULL 0.0001 units box
fix 1 movable nvt temp 300 300 1 drag 1
dump calcite all custom 200 dump.tensile.lammpstrj id mol type q xs ys zs fx fy fz
run 10000
However, on first try, the simulation will keep running (2 days for 10ps simulation) and eventually ends with following message:
“Error on procs…Bonds atom missing…”
Someone told me that it might be because the box has a large fraction of vacuum, so at the beginning of the script, I can add the processor command to specify 1 core in z-direction to reduce the instability issue:
processors * * * grid twolevel 32 * * 1
The simulation can run normally now, but with following video output and simulation error:
ERROR: Lost atoms: original 4800 current 3602
Appreciate if anyone know what is the error in my script… Thanks.