Dear lammps experts,
I use the reax this potential for Si O Cr,
but when I do this program
It has a wrong message for unknown parameter Reactive and stop
It is any problem about it
the file is below
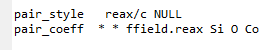
your pair_style command is incorrect for the reax/c substyle
your pair_coeff command is incorrect for the reax/c substyle
in fact, your final pair_coeff line will wipe out all previous ones, so it makes no sense to use hybrid at all since you would be treating all atoms with reax/c
your reax force field file seems to be broken, possibly due to editing in a text editor that applies (unwanted) line wrapping
Dear Axel:
I try to debug for the potential bug and wrap them to the correct site.
and change the pair_style and pair_coeff ,
but when I return to run the lammps
It has wrong message for “Warning :changed valency_boc for ov/un;val1;n.u” and “Warning : changed valency_boc for -2.8983”
And then I try to do the message but it doesnt work and lammps stop for running.
It that the potential file has a bug or I have a wrong step?
most likely, you made a mistake somewhere.
the message about changed valency is just a warning about an unused dummy entry in the force field file. it shows in many such files and has nothing to do with your simulation not completing.
the most common mistake with reaxff is to use the wrong units setting.