Dear LAMMPS users,
I am interested in calculating self-diffusivity of water in a porous solid structure using Velocity Autocorrelation Function.
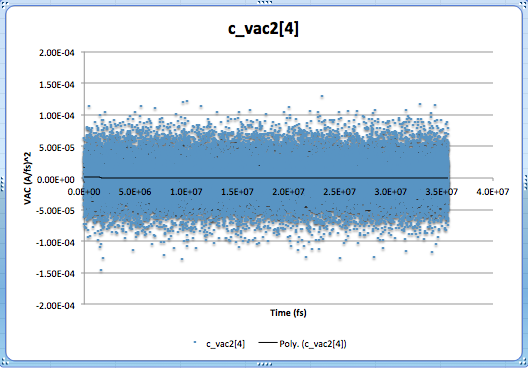
I have used “Fix Ave/time” to output the data calculated from "Compute VACF”. However, the VAC results obtained in this way do not make sense! As illustrated in the attached file, VAC values fluctuate continously around zero even after 35 ns. I wonder if using Fix Ave/time is the correct way of doing this?
Here is the way I have configured my input script:
group adsorbent molecule 24 # the solid structure
group sorbate molecule <= 23 # all fluid molecules
group oxygen type 1 # all central H2O atoms
……
compute vac oxygen vacf
compute vac2 sorbate vacf
……
fix 11 oxygen ave/time 1 1000 1000 c_vac[1] c_vac[2] c_vac[3] c_vac[4] file vacf1.dat
fix 12 sorbate ave/time 1 1000 1000 c_vac2[1] c_vac2[2] c_vac2[3] c_vac2[4] file vacf2.dat
fix 13 oxygen ave/time 1 1 1000 c_vac[1] c_vac[2] c_vac[3] c_vac[4] file vacf3.dat
fix 14 sorbate ave/time 1 1 1000 c_vac2[1] c_vac2[2] c_vac2[3] c_vac2[4] file vacf4.dat
By the way, in order to make sure that I have the right dynamic in my system, I have calculated self-diffusivity of the same system from MSD data successfully.
Your response is greatly appreciated.
Regards,
Amir