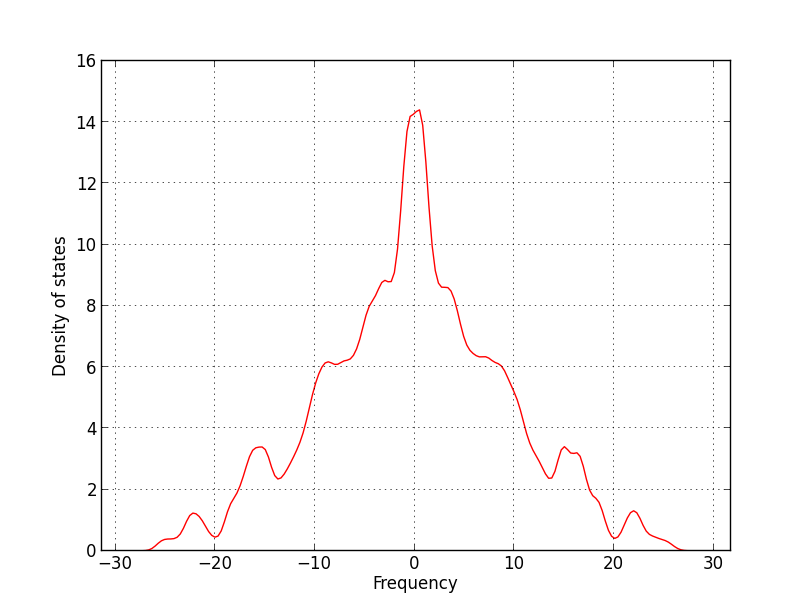
I am trying to use ReaxFF to calculate the vibrational spectra of a mineral, Ettringite. I tried to obtain the Dynamical Matrix from the forces in the lammps dump file, also, on the REAXFF trajectory file (defined on the reaxff control file). Unfortunately, the obtained spectra is wrong, with many negative frequencies, as you can see on the attached image. I have tried many kinds of minimizations, but none was successful. But, when I use the CLAYFF regular classical potential, the spectra is fine, with similar minimization. I don’t know where else to look, maybe I need to do something different to obtain the forces from REAXFF, anyone has ever faced this problem before?
ReaxFF parameterizations tend to have narrow applicability. Each parameterization is designed to reproduce a “training set” of data generated using higher order methods (DFT). If it wasn’t parameterized to reproduce the data you’re gathering, there’s no guarantee it will.
Hi Sylvia,
which set of reaxFF parameters are you using for ettringite?
if it’s ours [J Phys Chem A. 2012 Apr 19;116(15):3918-25], you need to know that we normally don’t train reaxFF force fields ‘directly’ from first or second derivatives of the energy (and we did not do so for ettringite), albeit one could argue that forces should (ideally) follow from accurate sampling of relative energies in cold compression curves, and other QM data we do train against.
We can certainly use explicit forces directly to train reaxFF potentials (e.g. using GARFfield), so you could in principle and if needed, re-optimize the parameter set to improve the dynamical matrix from forces for ettringite.
But, I would have to ask you if you have a pressing reason to use reaxFF for this. A reaxFF PES is not as smooth as a classical harmonic-type potential, and you’re 2nd derivatives might not be that well defined (i.e. wrong curvatures). Furthermore, you would need to make sure the forces are dumped at much higher frequency to avoid transient changes in sign (aliasing) coming from any non-harmonicities.
Have you tried calculating the vibrational spectrum from the velocity autocorrelation function (VAF) of your trajectory instead? you should be able to do so, but again you would have to make sure the dumps are done at sufficiently high frequencies to avoid aliasing problems, (i.e. this leads to very large dumps).
Best