Dear lammps user,
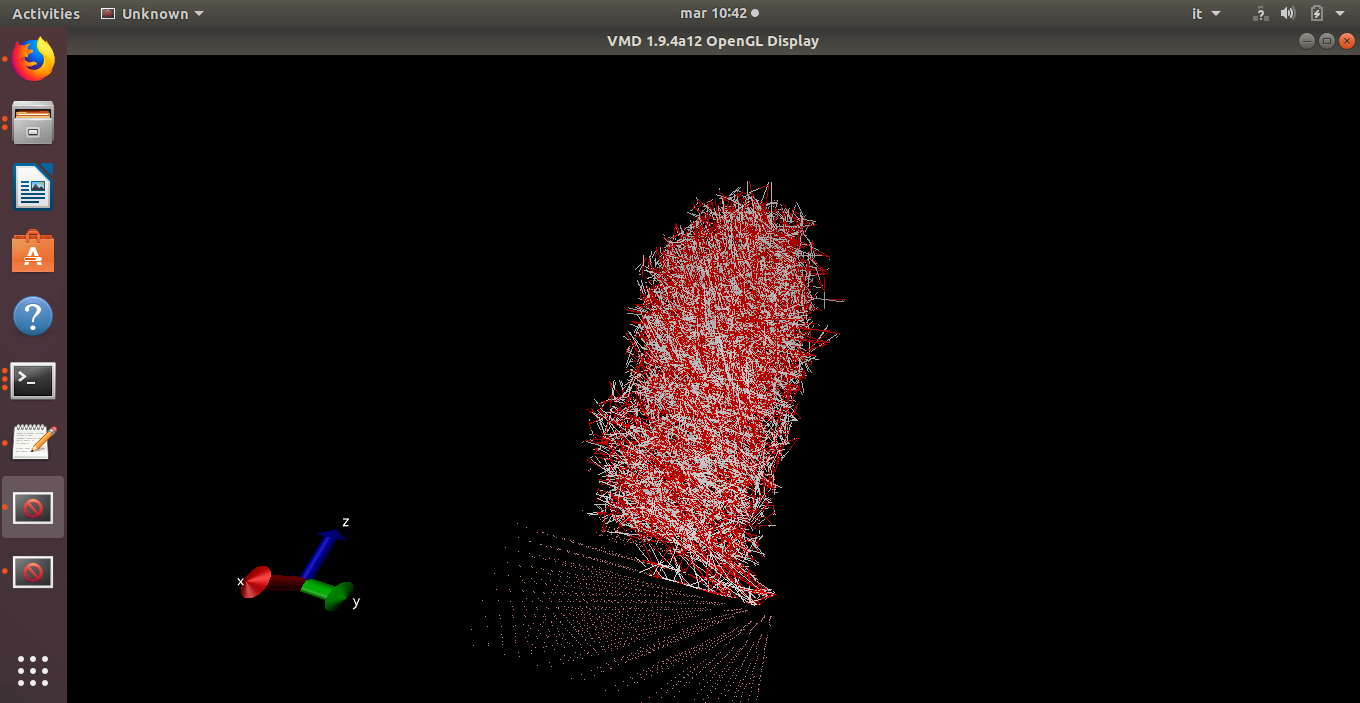
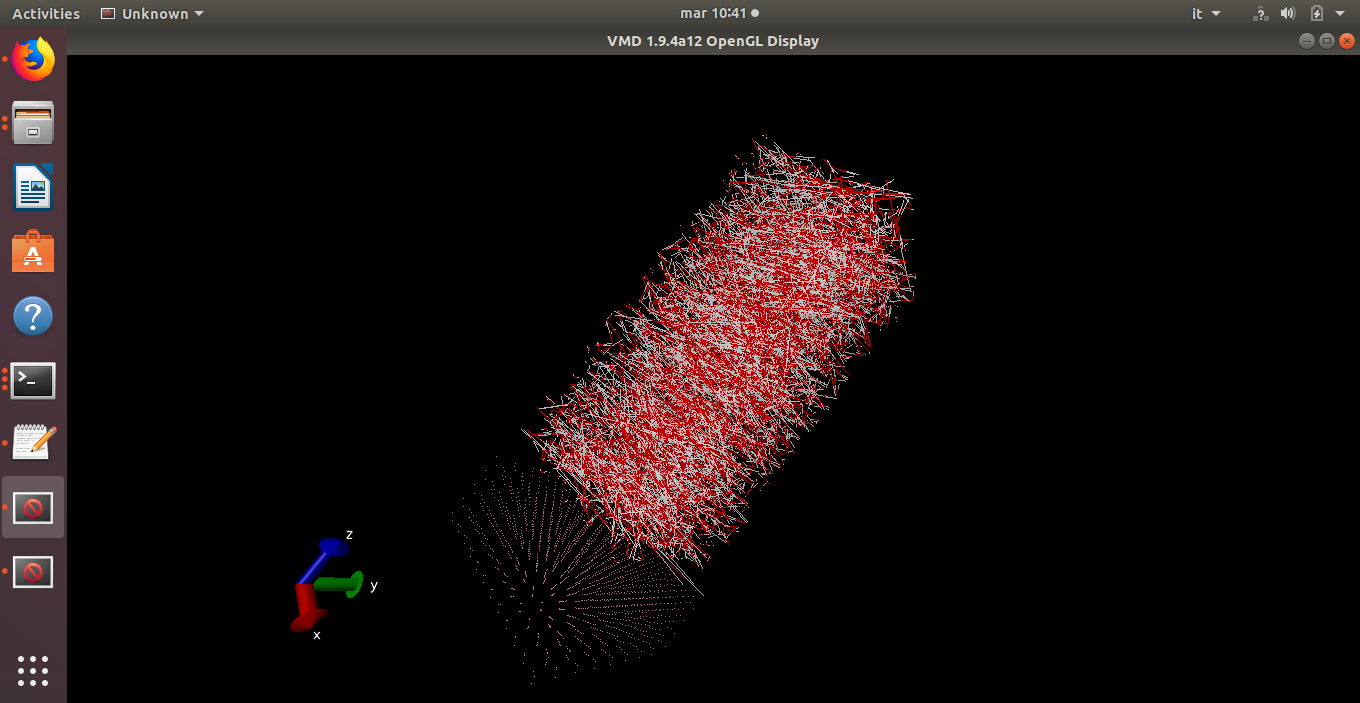
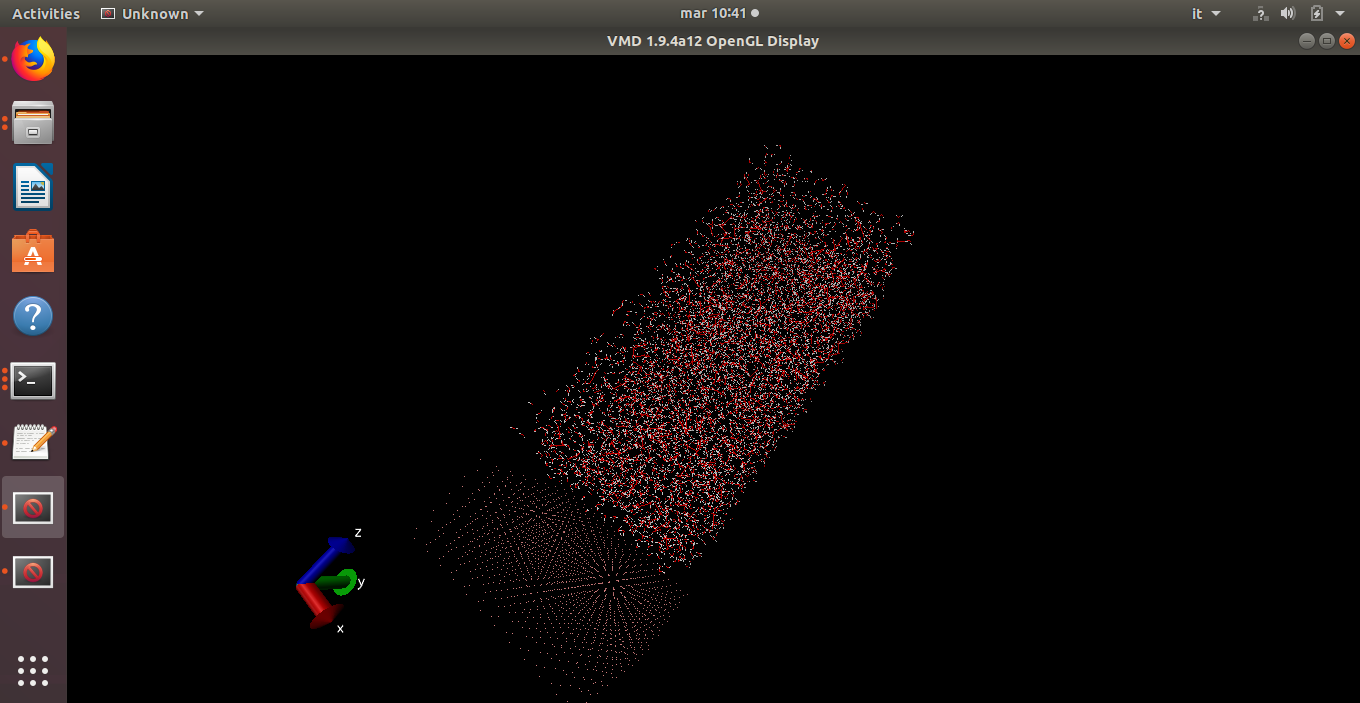
I’m having the following issue, i built my system with avogadro (Ge slab) and packmol to pack the box with water. Minimization seems to work but as soon as i try to thermalize i see bonds stretching and particles moving crazily.
This is the output, which seems to me almost meaningful:
Setting up cg style minimization …
Unit style : metal
Current step : 0
Per MPI rank memory allocation (min/avg/max) = 134.7 | 134.7 | 134.7 Mbytes
Step Temp KinEng Volume Lx Ly Lz Press E_pair E_vdwl E_mol TotEng E_long E_tail
0 0 0 1000000 50 50 400 159739.63 17460.166 17454.541 0.0091634717 17460.175 -18979.158 -2.1619983
99 0 0 1000000 50 50 400 269.92826 -8246.4694 -6336.2896 4.1266117 -8242.3428 -19071.408 -2.1619983
Loop time of 42.0237 on 1 procs for 99 steps with 16900 atoms
99.7% CPU use with 1 MPI tasks x no OpenMP threads
Minimization stats:
Stopping criterion = energy tolerance
Energy initial, next-to-last, final =
17460.1753297 -8241.55412973 -8242.34278974
Force two-norm initial, final = 9262.94 28.7378
Force max component initial, final = 702.425 4.08492
Final line search alpha, max atom move = 0.011189 0.045706
Iterations, force evaluations = 99 170
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total