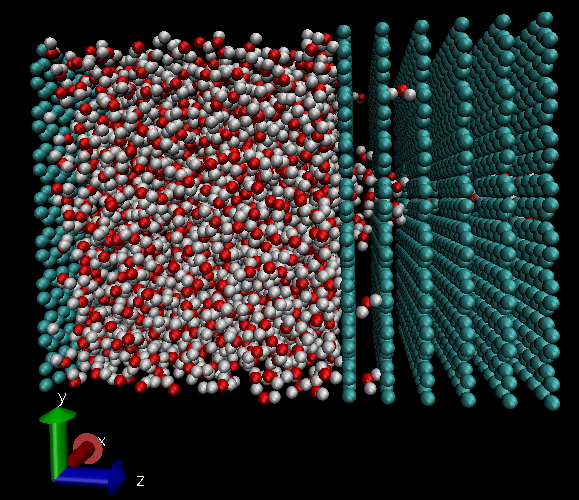
Dear Users and Developers,
I am simulating water transport through Multilayer graphene. I am applying DPD thermostat to fix temperature at 300 K. To perform non-equilibrium simulation I applied an external force on water molecules just in Z-direction. I also considered the graphene layers as fixed components (using fix setforce and fix move linear commands). After a short running I surprisingly found that water molecules are going out of the simulation box from between the graphene layers in Y direction. I dont know why?
Might it come from the periodic boundary condition (p p p) that I have set?
Please find the attached to take a look at the model.
My input script
units real
dimension 3
boundary p p p
atom_style full
read_data LAMMPS-DaTa.dat
group carbon type 1
group hydrogen type 2
group oxygen type 3
group water type 2 3
set group oxygen charge -0.8476
set group hydrogen charge 0.4238
set group carbon charge 0.0000
comm_style brick
comm_modify vel yes
pair_style hybrid/overlay lj/cut/coul/long 10.0 10.0 dpd/tstat 300.0 300.0 10.0 34387
pair_coeff 1 1 lj/cut/coul/long 0.000000 0.0000
pair_coeff 1 2 lj/cut/coul/long 0.00000 0.00000
pair_coeff 1 3 lj/cut/coul/long 0.093627 3.1900
pair_coeff 2 2 lj/cut/coul/long 0.000000 0.0000
pair_coeff 2 3 lj/cut/coul/long 0.000000 0.0000
pair_coeff 3 3 lj/cut/coul/long 0.155300 3.16900
pair_coeff 1 1 dpd/tstat 0.0 0.0
pair_coeff 1 2 dpd/tstat 0.0 0.0
pair_coeff 1 3 dpd/tstat 0.02 10.0
pair_coeff 2 2 dpd/tstat 0.0 0.0
pair_coeff 2 3 dpd/tstat 0.0 0.0
pair_coeff 3 3 dpd/tstat 0.02 10.0
bond_style harmonic
bond_coeff 1 529.581 1.0
angle_style harmonic
angle_coeff 1 37.95 109.47
fix force_zero carbon setforce 0.0 0.0 0.0
fix vel_zero carbon move linear 0.0 0.0 0.0 units lattice
velocity water create 300.0 34387 rot yes dist gaussian # for water
kspace_style ewald 1.0e-6
*************** Setting ******************************
neighbor 3.0 bin
neigh_modify delay 0 every 10 check yes
reset_timestep 0
delete_atoms overlap 0.3 all all
#minimize 1.0e-4 1.0e-6 100 100000
#min_style cg
compute peatom all pe/atom
compute keatom all ke/atom
thermo 100
thermo_style custom step temp pe etotal press vol
thermo_modify norm no flush yes
fix ensemble_set water nve #temp 300.0 300.0 100.0
fix spce_model water shake 0.0001 20 0 b 1 a 1
fix 3 water addforce 0.0 0.0 0.215 # g=0.05
variable end equal 10000
dump pos all custom ${end} pos_filename id element type q x y z
dump_modify pos format “%5d %5s %d %13.10f %17.12f %17.12f %17.12f”
dump_modify pos sort id
dump_modify pos element C HW OW
dump vel all custom ${end} vel_filename id element vx vy vz
dump_modify vel format “%5d %5s %18.15f %18.15f %18.15f”
dump_modify vel sort id
dump_modify vel element C HW OW
#dump trj1 all atom 100 wat.trj
dump trj all custom 100 wat.dat id element type x y z vx vy vz fx fy fz
dump_modify trj sort id
dump_modify trj element C HW OW
write_data waterinfo.data
write_restart waterinfo.restart
timestep 0.05
run 10000