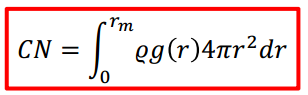Please direct all questions to the LAMMPS mailing list.
Short answer: column 3 gives the integral, not the integrand.
Long answer: The third column does indeed calculate the quantity CN(r_m) given in your equation, which is equal to the integral on the right side (not the integrand, as your comment suggests). If you are still not getting that, you are probably doing some kind of filtering (by using a group other than group all, or by selecting only certain atom type pairs). To check this, I suggest you try out the compute rdf in a simple LJ simulation with only one type.
I made a mistake in my previous question. I wanted to say the 4th column of the output of compute_RDF should be proportional to g®*r^2dr that g® in column 3 of the output but it is not!!! (according to what we see in the link you sent in wikipedia)
I made a mistake in my previous question. I wanted to say the 4th column
of the output of compute_RDF should be proportional to g(r)*r^2dr that g(r)
in column 3 of the output but it is not!!! (according to what we see in the
link you sent in wikipedia)
I would appreciate if I have your further advice.
if you believe that LAMMPS is incorrect, you have to provide *convincing
proof*.
saying, that you do not get the expected result in your calculations proves
nothing in that respect.
why don't you just follow aidan's suggestion and set up a simple
calculation where the g(r) and the coordination number is known and compare
that to what compute rdf provides as well as your calculation.