Dear all lammps users:
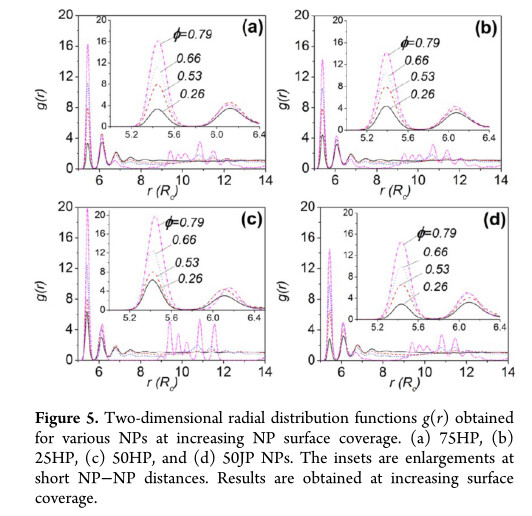
I want to use lammps to calculate the two-dimensional radial distribution function of nanoparticles on the oil-water interface. I mainly want to obtain the nearest distance between particles from the abscissa of the first peak of the two-dimensional radial distribution function, as shown in the following figure.
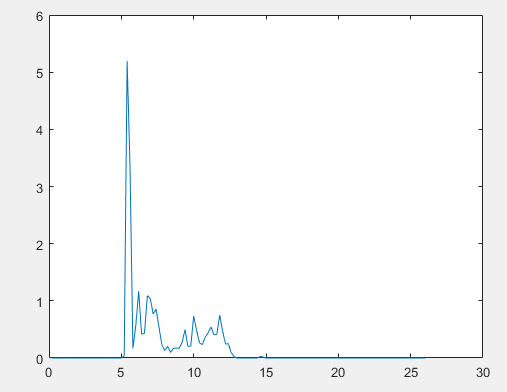
But lammps calculates the three-dimensional radial distribution function, so I directly use lammps to output the coordinates of the central atom of particles, and then import the coordinates into Matlab to calculate the RDF of the central atom, However, the central atom at the side of the simulation box will not be processed, resulting in the inaccurate calculation of 2Drdf,as shown in the following figure. What can be done to solve it?