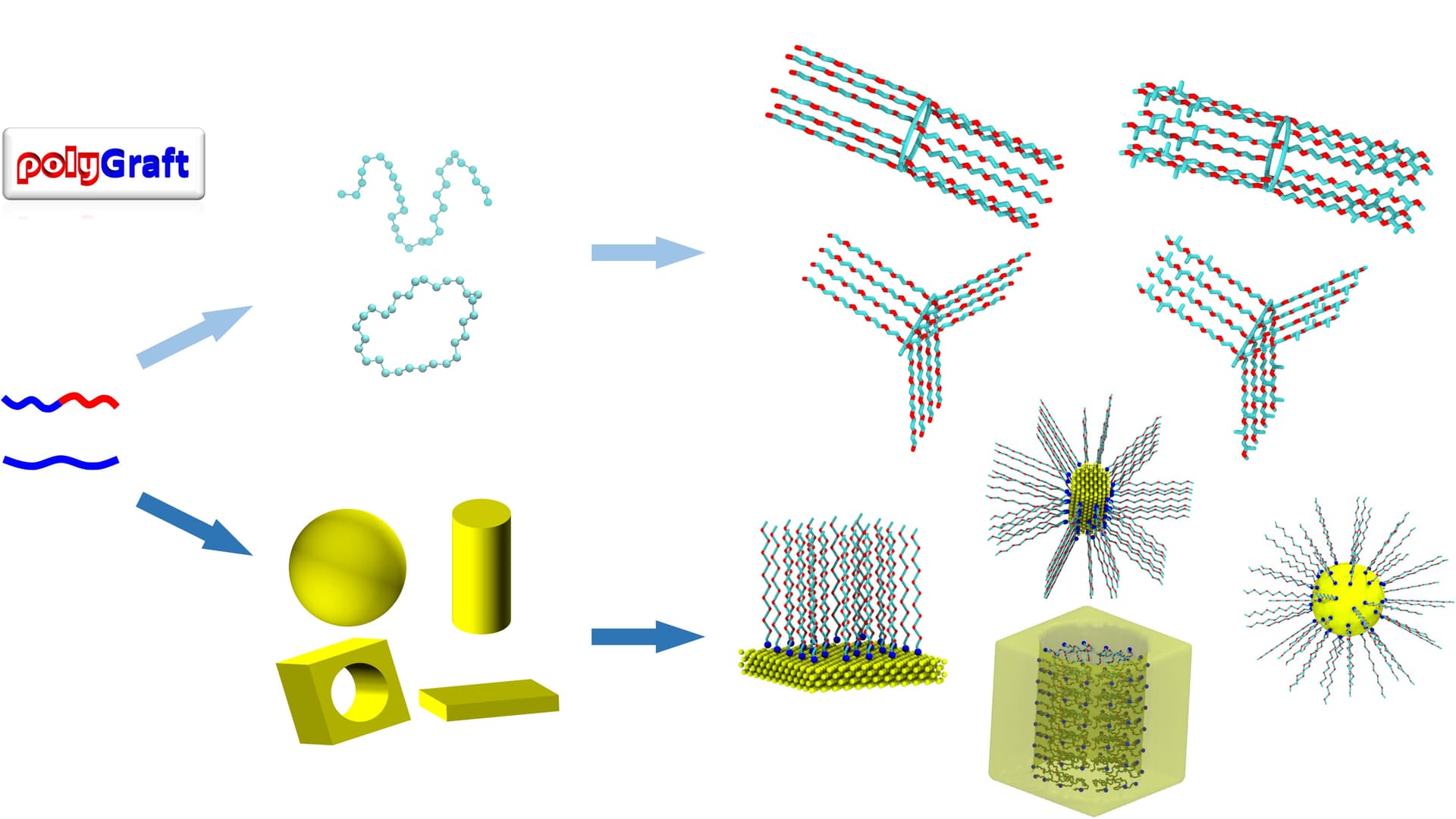
Here is a Python package, for the generation of molecular structure and topology of polymer-grafted hybrid materials, including grafting polymer to planar, sphere, and in (pore) and out of a cylinder.
Thanks for introducing this tool, it fits an excellent and growing research field.
Have you tried any particular conversion tools with your package’s output, and can you recommend any?
As someone who has experience with both GROMACS and LAMMPS I don’t think it’s straightforward to convert between them. GROMACS relies heavily on “fragment” templates to specify topology (which makes sense given the protein focus) which are not used much in the LAMMPS paradigm. Also, GROMACS inputs tend to wrap bond topologies with force field parameters – in LAMMPS inputs bonds, angles and dihedrals are read from the data file, but the force field functionals are given in the script (and coefficients can go in either place).
So it might make sense to develop a dedicated LAMMPS file generator together with your package. I am happy to provide help as needed.
Thanks. And it’s simple: move (translate and rotate) a reference polymer (or possibly from multiple polymers if, e.g., a bimodal distribution or Janus-type grafting is of interest) to the desired locations, and one gets the geometry. The system topology needs just translation on the reference polymer, and deals with the cross term between grafts and substrates.
There are indeed many differences in software philosophy between LAMMPS and Gromacs. But here, no force field parameters are provided by the package. Here the topology simply refers to the connection of points (atoms) between the graft atoms, substrate atoms, and substrate-polymer (e.g., bond, but one can add angle if needed). While the bonded interaction (function types and parameters) has to be set by the user.
Anyway, thanks for the suggestion. Let’s wait and see if the other packages (as suggested by Axel) work smoothly. If needed, I’ll be happy to work with anyone interested in that. Any questions or suggestions are welcome to the GitHub repo (it may be more convenient to discuss there).
Update: lammps data file for the graft and substrate can be directly used/read for generation and the generated structure can be output in lammps data file.