I want to calculate the interactions between two groups of atoms. One is carbon atoms with diamond structure without giving any potentials, the other one is SiC with Tersoff potential. And the interactions between these two groups are represented by Morse potential. I read the manual that compute group/group cannot be used for pairwise potential, so does that mean I cannot use this command? And also is there any other commands to deal with this problem?
Thank you very much for your reply sir. Yes, I thought it may not tell me if this command is not suitable for my system. I actually have got the result without any errors. So I think it should be working well. It’s just the values I obtained from this command don’t look correct, and that brought me this question. Now I think it’s probably something else that is wrong and I’ll check it. Thank you for your help!
Sorry about that sir. I didn’t tell it at the beginning because I feel like it’s not really a LAMMPS command problem if it’s not due to that compute group/group.
So I want to calculate the interactions between two groups of atoms. One contains carbon atoms with diamond structure as the indenter, the other one is the debris generated during the indenter scratching on the SiC/graphene workpiece. (Here the position of graphene layers are very low so that it’s not considered in this case.) That is to say the debris are SiC. I set up dynamic group to the debris since the number of atoms would increase with the scratching. I don’t define any potential for the diamond indenter to treat it as a hard tool, and Tersoff potential is used for SiC, Morse potential for the interactions between SiC and the indenter. I attached the input file here so that you can look at the details about the configurations. in.create (5.9 KB)
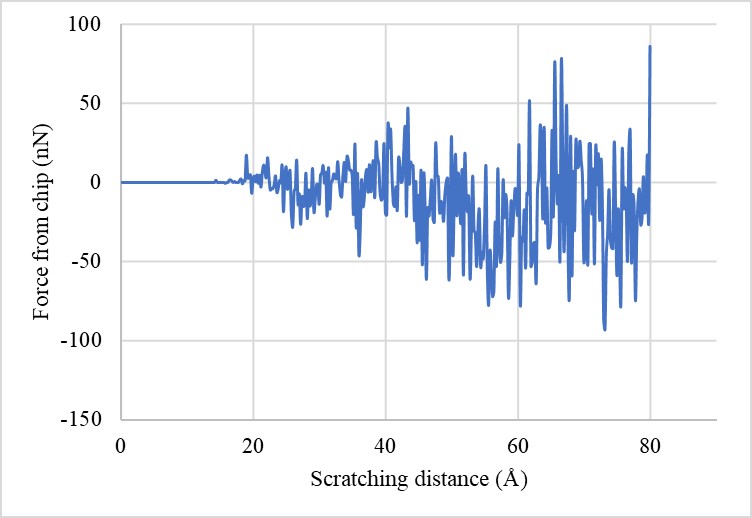
The issue I meet is that I tried to use “compute 6 chips group/group tool” to calculate the component force from the tool to the debris along the scratching direction, which is the positive direction of x-axis. The output is c_6[1]. I analyze in the end I should be getting the component forces that are all positive values with the scratching distance increased since the indenter is pushing the debris along the positive direction along x-axis. However, in the results I have many negative values about that force. I also attached result figure here.
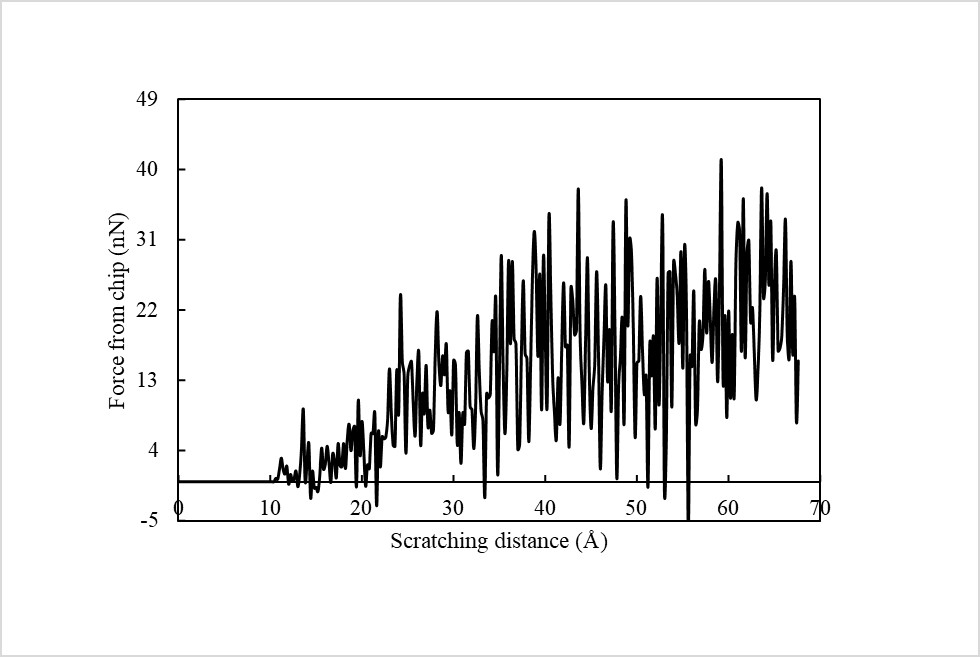
The final result looks better, though there are still some negative values. I don’t know why there are negative values for the force and I thought it may be related to the compute group/group command. Thank you very much!
Sorry, but that makes even less sense than not mentioning it at all.
Whether you get the expected result or not is always either an issue with the implementation, the model, or your expectations or a combination of those. However, if you do get the results you expect, that does not automatically mean that your model is correct or your results are correct.
So, before you can trust your results and your model, you need to construct simpler scenarios and consult with people that have experience in similar calculations and confirm that both your model and your approach are suitable. However, figuring this out is - as you expected - beyond the scope of LAMMPS. However, not mentioning that you already did a calculation and did not get the expected results and that you are trying to figure out where the problem could be, is a valid LAMMPS question.
Or in short, the first two lines of your response are on topic, the rest not.
Here is the main problem: You need to think about what it means that an (instantaneous!) force may become negative, especially when you have different materials. I don’t think it is that surprising. It is a very common mistake to assume that either your input is wrong or your software is not working correctly, when it is more likely that you have not thought through what your model means and how it works at the atomic level. You cannot simply assume behavior as if this was a bulk material.
Thanks for your reply sir. Yes, I think your comment really helps me reconsider my system and what will the atoms behave in the nano level. Although the net force is negative, but it may still be reasonable because it’s not in macro scale. The important thing is to make sure that the simulation, the model or the system follows the basic laws and is reasonable. Thank you very much sir!