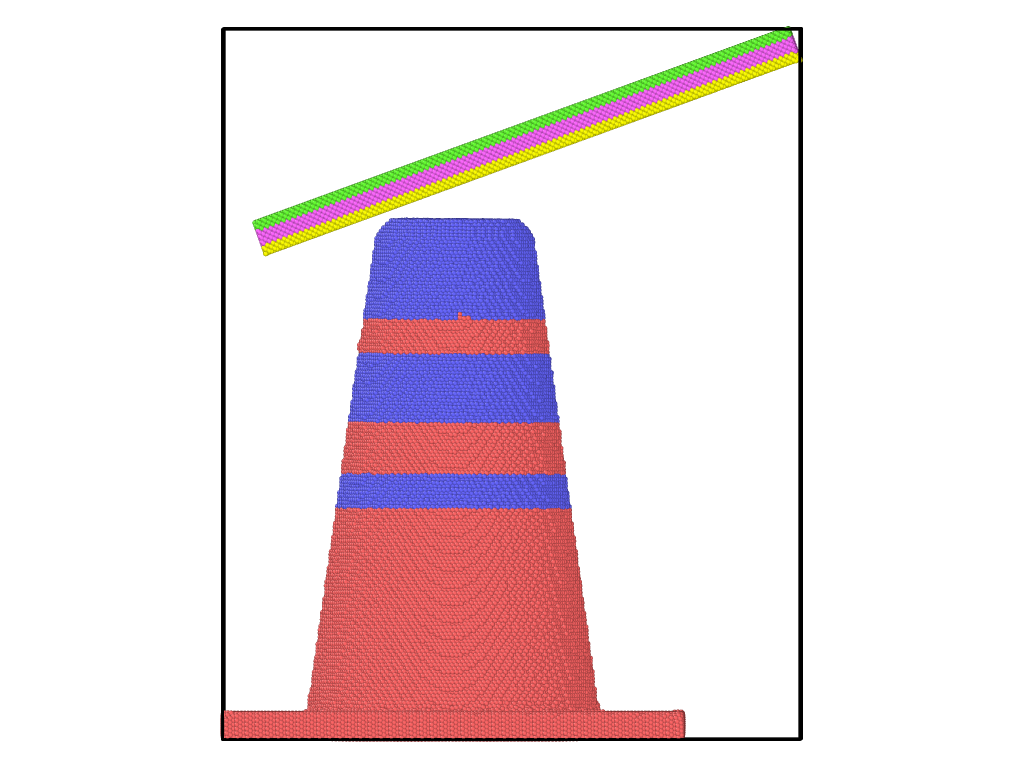
Above is the image
I want to make the slab down to do the compress test and measure the stress
the green region is setforce 0 0 0 and the value of stress is obtained from the yellow region of the slab
and the yellow region, the pick region and the Pillar part are set 300K by fix berendsen
the thermostatting process is OK
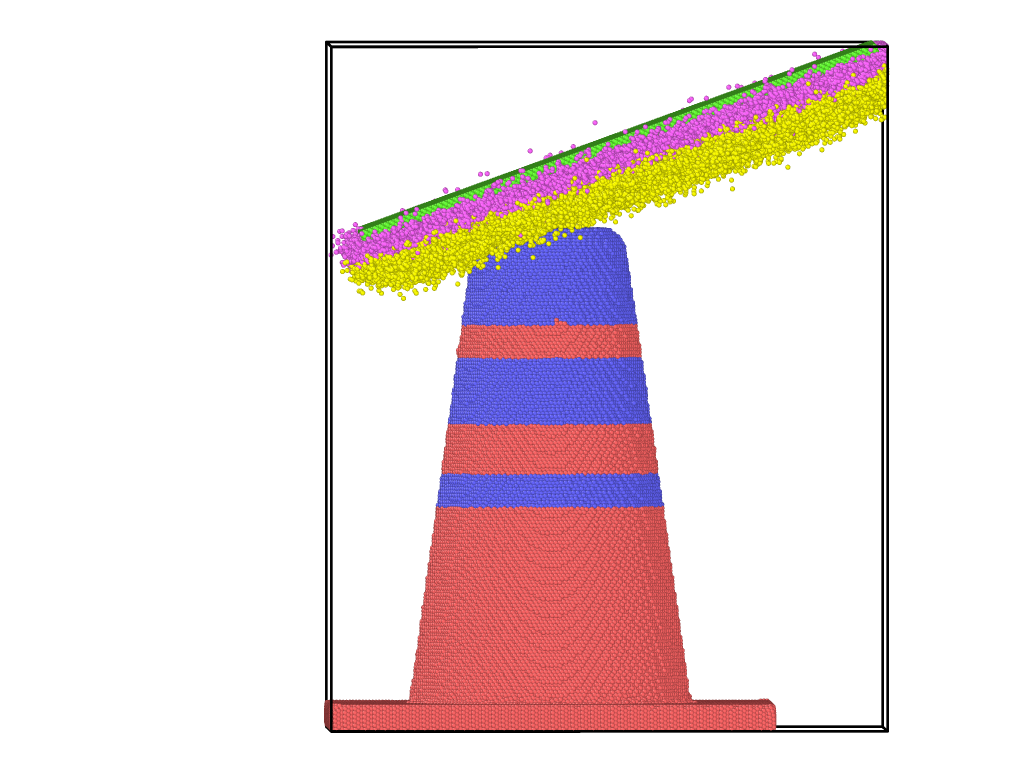
but I got this error: ERROR on proc 440: Too many neighbor bins (…/nbin_standard.cpp:184) as soon as it went into the slab-downward process
error message is like below:
Total # of neighbors = 0
Ave neighs/atom = 0.0000000
Neighbor list builds = 2000
Dangerous builds = 1980
WARNING: Temperature for fix modify is not for group all (…/fix_nh.cpp:1426)
WARNING: One or more atoms are time integrated more than once (…/modify.cpp:287)
Setting up Verlet run …
Unit style : metal
Current step : 421468
Time step : 0.002
Per MPI rank memory allocation (min/avg/max) = 10.10 | 10.90 | 12.45 Mbytes
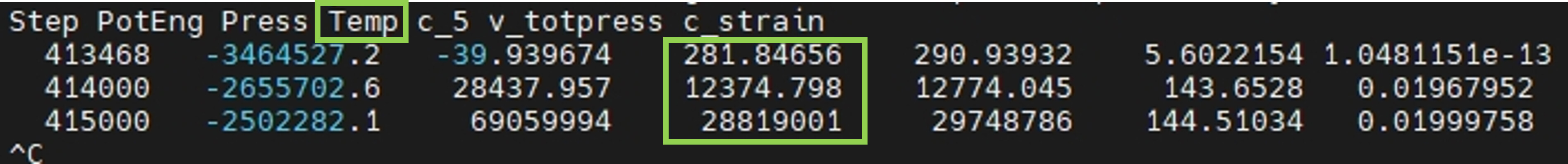
Step PotEng Press Temp c_5 v_totpress c_strain
421468 -3465529.7 -130.28159 268.40814 277.06381 5.5061556 0
ERROR on proc 0: Too many neighbor bins (…/nbin_standard.cpp:184)
ERROR on proc 1016: Too many neighbor bins (…/nbin_standard.cpp:184)
ERROR on proc 844: Too many neighbor bins (…/nbin_standard.cpp:184)
My input is like this:
#-----initialization-----
units metal
dimension 3
atom_style atomic
timestep 0.002 #---------------slab--------------
group top3 type 3
group top4 type 4
group top5 type 5
group toppp union top3 top4 top5
group nvtpart union wtf_1 wtf_2 wtf_3 wtf_4 wtf_5 wtf_6 top3 top4
change_box all boundary s s f #change boundary #-----mass-----
mass 1 63.55
mass 2 63.55
mass 3 12.0096 #carbon mass (amu)
mass 4 12.0096
mass 5 12.0096 #------ force field ------
pair_style hybrid eam/alloy tersoff lj/cut 3.615
pair_coeff * * eam/alloy Cu_zhou.eam.alloy Cu Cu NULL NULL NULL
pair_coeff * * tersoff SiC.tersoff NULL NULL C C C
pair_coeff 12 3 lj/cut 0.001034 2 3.615
pair_coeff 12 4 lj/cut 0.001034 2 3.615
pair_coeff 12 5 lj/cut 0.001034 2 3.615 #------------computation--------------
compute 5 nvtpart temp
compute peratom all pe/atom
compute 1 top3 stress/atom NULL stress(volume/atom)
compute 2 top3 reduce sum c_1[1] c_1[2] c_1[3] stressvolume
variable totpress equal -(c_2[1]+c_2[2]+c_2[3])/(3(3452105))/10000 #This is what I need, which is stress(GPa)
Too many neighbor bins typically happens when your simulation box becomes too large.
In your case, that can happen as soon as your two objects get into contact and then atoms may detach. Once those atoms move away, there is nothing to stop them and because you have shrinkwrap boundaries, the box will grow. This effect is particularly rapid, when the two object collide at a (macroscopically) high speed. This can be easily validated by visualizing the resulting trajectory and monitoring the box dimensions.
Possible approaches to avoid the issue (you may need a combination of multiple of those, some have a significant performance impact, others may change the physics):
use smaller impact velocities
use shorter time step
use fixed boundaries and either ignore lost atoms or add a harmonic/reflecting wall
use periodic boundaries
use a large bin size for neighbor lists with neigh_modify
use nsq neighbor list construction
You seem to be running a very large system with an equally large number of MPI processes. I would start experimenting with a much smaller system so you can run on a local machine to figure out the possible changes to your simulation more quickly and conveniently.
Thanks for your advice.
I try the methods you mentioned
include
use smaller impact velocities(-0.15 to -0.075)
use a shorter time step(0.002 to 0.001)
ignore lost atoms
use nsq neighbor list construction(2.0 nsq)
it works, but the temperature of the system increases dramatically.
how can I fix this by adjusting the physics setting like temperature control object
Your system’s potential energy is changing by millions of eV per picosecond, which shows that under the parameters you’ve chosen, the process you’re simulating is extremely disruptive.
You could brute-force the system’s total kinetic energy to stay constant using a Gaussian isokinetic thermostat (fix nvk), but that does not change the unphysically high energy changes in your system. If you replace a physically-wrong simulation at one million Kelvin with a physically-wrong simulation at 300K you have not gained anything.
At the very minimum, you need to find a stable integration timestep – that is, one where over at least a thousand timesteps the energy change rate (in units of energy per time, not per timestep) does not consistently vary with the size of the timestep.
Also, you should post your full script. The log you posted has a warning about multiple integration, but I don’t see where that comes from in the script you posted, which means the thing destroying your simulation might be something we can’t see and therefore something we can’t help with. Please post your script using backticks,
In particular, in units metal, a speed of 0.15 to 0.75 angstroms per picosecond is (in more familiar units) between 54 to 270 kilometres per hour. These are highway driving speeds, not compression stress-strain testing speeds, and I think the sudden and irreversible destruction of the nanoindenter is actually a pretty physically sensible outcome.
#-----initialization-----
units metal
dimension 3
atom_style atomic
read_restart 0_totally-keeptemp.restart
fix 5 all dt/reset 5 NULL 0.001 0.5 emax 30 units box
neighbor 2.0 nsq
variable scale equal 1.0
variable tdamp equal 10
variable file string 0_
variable theta equal 20*3.1415926/180
variable z_scale equal 375*${scale}
variable z_1 equal ${scale}*124.5
variable z_2 equal ${scale}*145.5
variable z_3 equal ${scale}*175.5
variable z_4 equal ${scale}*217.5
variable z_5 equal ${scale}*240
variable z_6 equal ${scale}*380
variable low_1 equal 105*${scale}
variable low_2 equal 87.9*${scale}
variable low_3 equal 168.54*${scale}
variable low_4 equal 250.02*${scale}
variable low_5 equal 332.34*${scale}
variable low_6 equal 415.5*${scale}
variable high_1 equal 45*${scale}
variable high_2 equal 45.9*${scale}
variable high_3 equal 42.54*${scale}
variable high_4 equal 40.02*${scale}
variable high_5 equal 38.34*${scale}
variable high_6 equal 37.5*${scale}
variable cone_hi equal 300*${scale}
region model_1 prism -${z_scale} ${z_1} -140 140 -375 375 0.0 0.0 0.0 units box
region model_2 prism ${z_1} ${z_2} -140 140 -375 375 0.0 0.0 0.0 units box
region model_3 prism ${z_2} ${z_3} -140 140 -375 375 0.0 0.0 0.0 units box
region model_4 prism ${z_3} ${z_4} -140 140 -375 375 0.0 0.0 0.0 units box
region model_5 prism ${z_4} ${z_5} -140 140 -375 375 0.0 0.0 0.0 units box
region model_6 prism ${z_5} ${z_6} -140 140 -375 375 0.0 0.0 0.0 units box
group wtf_1 region model_1
group wtf_2 region model_2
group wtf_3 region model_3
group wtf_4 region model_4
group wtf_5 region model_5
group wtf_6 region model_6
change_box all boundary s s s #change boundary
#---------------slab--------------
group top3 type 3
group top4 type 4
group top5 type 5
group toppp union top3 top4 top5
group nvtpart union wtf_1 wtf_2 wtf_3 wtf_4 wtf_5 wtf_6 top3 top4
change_box all boundary s s f #change boundary
#-----mass-----
mass 1 63.55
mass 2 63.55
mass 3 12.0096 #carbon mass (amu)
mass 4 12.0096
mass 5 12.0096
#------ force field ------
pair_style hybrid eam/alloy tersoff lj/cut 3.615
pair_coeff * * eam/alloy Cu_zhou.eam.alloy Cu Cu NULL NULL NULL
pair_coeff * * tersoff SiC.tersoff NULL NULL C C C
pair_coeff 1*2 3 lj/cut 0.001034 2 3.615
pair_coeff 1*2 4 lj/cut 0.001034 2 3.615
pair_coeff 1*2 5 lj/cut 0.001034 2 3.615
#------------computation--------------
compute 5 nvtpart temp
compute peratom all pe/atom
compute 1 top3 stress/atom NULL #stress*(volume/atom)
compute 2 top3 reduce sum c_1[1] c_1[2] c_1[3] #stress*volume
variable totpress equal -(c_2[1]+c_2[2]+c_2[3])/(3*(345*210*5))/10000 #This is what I need, which is stress(GPa)
compute 3 top5 displace/atom #displacement(vector)
compute strain top5 reduce ave c_3[4] #translate into scalar
#-----------variable------------------
variable theta equal 20*3.1415926/180
variable file string 0_ia
variable vz equal -0.075
variable vx equal ${vz}*tan(${theta})
#----------------keep temperature------------
fix top top5 setforce 0.0 0.0 0.0
fix freeze down1 setforce 0.0 0.0 0.0
fix 1 all nve
fix 2 nvtpart temp/berendsen 300.0 300.0 10.0
fix_modify 2 temp 5
dump 8 all custom 1000 ${file}_keep*.dump id type xu yu zu c_peratom
thermo 2000
thermo_style custom step pe temp c_5
run 12000
unfix 2
undump 8
#----------------fix---------------
#fix 6 top1 ave/time 1 250 250 v_totpress
fix xwalls all wall/reflect xlo EDGE xhi EDGE
fix ywalls all wall/reflect ylo EDGE yhi EDGE
fix zwalls all wall/reflect zlo EDGE zhi EDGE
#-----------down-------------------
fix 2 nvtpart nvt temp 300.0 300.0 10
fix_modify 2 temp 5
velocity top5 set ${vx} 0.0 ${vz} sum yes units box
dump 8 all custom 1000 ${file}_def_*.dump id type xu yu zu c_peratom
thermo 1000
thermo_style custom step pe press temp c_5 v_totpress c_strain
thermo_modify lost ignore flush yes
run 400000
unfix 2
undump 8
restart 4000 ${file}_ia_*.restart
write_restart ${file}_def.restart
I read a restart file that has thermostatted top3,top4, and the pillar at 300K
Should I post the full script of that restart file? Cause I didn’t see a warning about multiple integration in it
above #-----mass----- , I just made atoms into groups(top5 and nvtpart, nvtpart including top3 top4 and pillar)
in #-------keep temperature-------- , I thermostatted nvtpart at 300K for a while
and in #-----------down------------- , I made the slab downward
Well you do have multiple integrators, because your fix 1 all nve is not unfixed before your fix 2 nvtpart nvt (so after that second fix, the nvtpart group is being integrated by both fix 1 and fix 2). That guarantees simulation problems. By the way, this is also why you shouldn’t reuse fix names – LAMMPS will let you do it, but you’re opening yourself up to all kinds of confusion when you or someone else has to subsequently read your script.
Additionally, all fix nvt instances will start their own temperature computes, so your fix_modify lines aren’t necessary – they don’t do any harm right now but the more unnecessary lines you have, the more chance you have of a typo causing an error when you do change your script.
I don’t know your exact field or model, so I can’t give specific advice (for example, in my field we almost always use the Nose-Hoover thermostat, but there are other applications where the Berendsen thermostat is perfectly fine for pre-equilibration). You need to know what a valid or invalid set of simulation results roughly looks like. If you see a clear problem with your results, we can often help you guess where that problem comes from; but we don’t know your field well enough to predict beforehand whether this parameter or that will work.