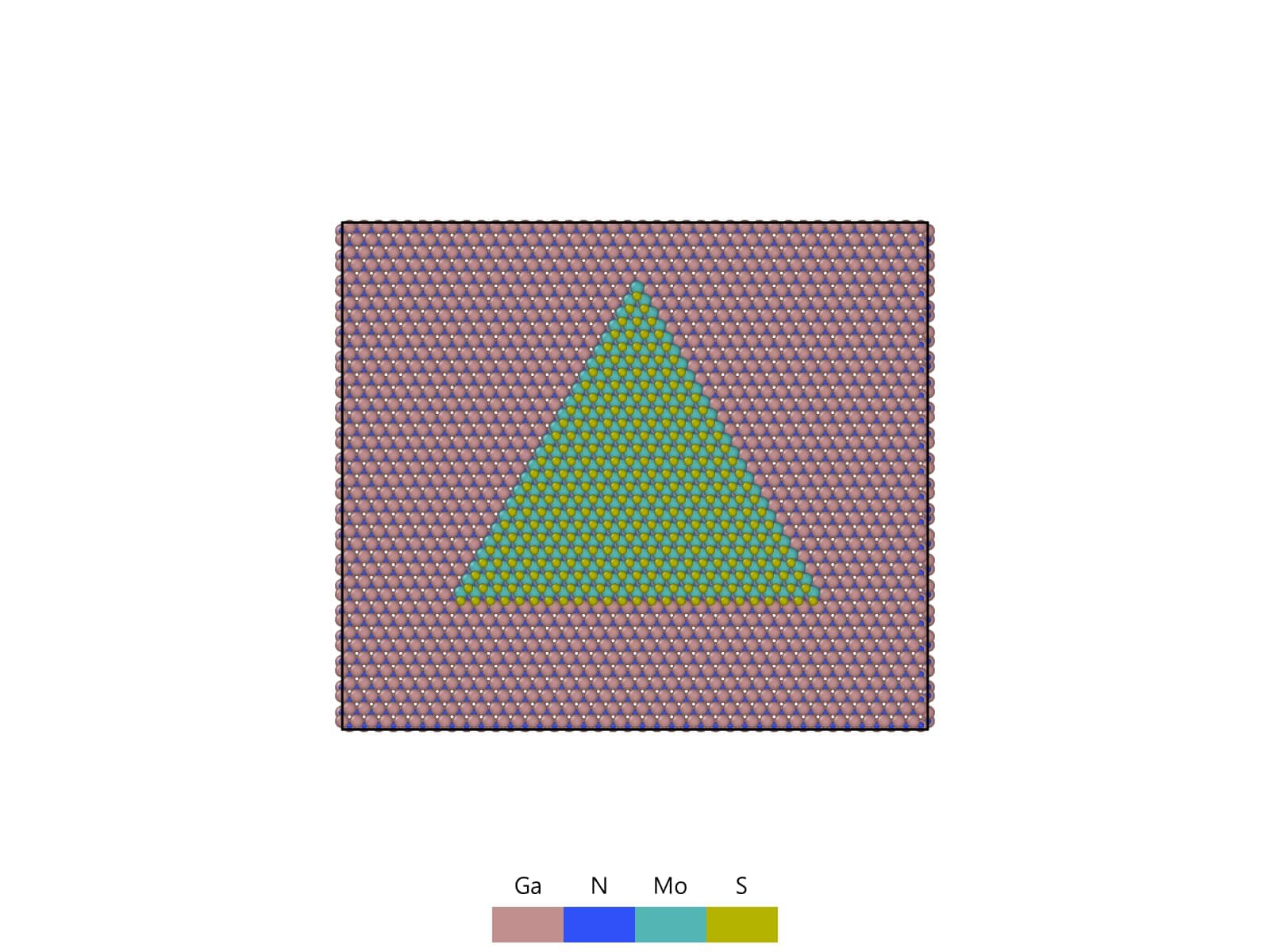
I am a beginner with LAMMPS and I would like to evaluate the strain in MoS₂ that is induced by lattice mismatch with a GaN substrate.
My LAMMPS version is 22 JUL 2025.
In my simulations I use a REBO potential for MoS₂, a Tersoff potential for GaN, and Lennard-Jones (LJ) cross-interaction terms parameterized from UFF for the MoS₂–GaN interface.
I place a monolayer MoS₂ triangular flake on three different GaN facets, (0001), (10-10), and (10-11), and run NVT simulations.
The simulations are performed first at 0.01 K, and then with a temperature ramp from 0.01 K to 300 K in NVT, in order to heat the system slowly and prevent MoS₂ from detaching from the GaN surface. From the dump files I post-process the trajectories to compute the Green–Lagrange strain tensor and compare the resulting strain in MoS₂ for each GaN facet.
Based on a simple lattice-mismatch analysis, I would expect MoS₂ on the (10-10) and (10-11) facets to experience stronger tensile strain than on (0001). However, in my simulations, the MoS₂ shows the largest tensile strain when it is placed on the GaN (0001) facet.
My question is:
Is this a reasonable way to calculate the lattice-mismatch-induced strain in MoS₂ using LAMMPS?
Is there an obvious problem or conceptual flaw in my setup?
In my opinion, it seems that the interlayer interaction described by the LJ potential is not strong enough to hold the MoS₂ flake in a stretched configuration, so MoS₂ relaxes before a clear lattice-mismatch–induced tensile strain can be fully maintained.
variable Qtemp equal 0.01 # K
variable tstep equal 0.0005
variable tdamp equal ${tstep}*100
variable relax_run equal 6000000
#----------initialize--------------------------------------
units metal
atom_style full
boundary p p f
timestep ${tstep}
#--------read data-----------------------------------------
read_data 001.data
mass 1 69.723 # Ga
mass 2 14.0067 # N
mass 3 32.065 # S
mass 4 95.94 # Mo
group GaN type 1 2
group MoS2 type 3 4
#-------------FF-------------------------------------------
pair_style hybrid tersoff rebomos lj/cut 10.0
pair_coeff * * tersoff GaN.tersoff Ga N NULL NULL
pair_coeff * * rebomos MoS.REBO.set5b NULL NULL S M
# Cross pairs
pair_coeff 1 3 lj/cut 0.01462 3.750 10.0 # Ga-S
pair_coeff 1 4 lj/cut 0.00661 3.312 10.0 # Ga-Mo
pair_coeff 2 4 lj/cut 0.0026956 2.990 10.0 # N-Mo
pair_coeff 2 3 lj/cut 0.0059625 3.428 10.0 # N-S
# ---------- regions -------------------------------------
region bot block INF INF INF INF INF 4.509 units box
region top block INF INF INF INF 4.509 INF units box
#-----------group-----------------------------------------
group fbot region bot
group ftop region top
group GaN_fixed intersect GaN fbot
group GaN_mobile subtract GaN GaN_fixed
#-------------fix-----------------------------------------
fix f0 fbot setforce 0 0 0
#---------- neighbor settings ----------------------------
neighbor 2.0 bin
neigh_modify every 10 delay 0 check yes one 25000 page 250000
#----------minimize---------------------------------------
thermo_style custom step time temp pe etotal
thermo 1000
dump d0 all custom 1 before_minimize.dump id type x y z
run 0
undump d0
min_style cg
minimize 1.0e-8 1.0e-8 500000 500000
compute mytemp ftop temp
dump d1 all custom 1 after_minimize.dump id type x y z
run 0
undump d1
dump d1 all custom 10000 traj.dump id type x y z
#--------------NVT ensemble_Temperature_Rampup------------------------------
fix f1 ftop nvt temp ${Qtemp} ${Qtemp} ${tdamp}
run ${relax_run}
write_dump all custom final.dump id type x y z
@doyounglim15 Thanks you for posting a well laid out post with all required context and information and a properly formatted input deck. It would make our life much easier if everybody, and especially first time posters, would follow your example.
Unfortunately, your questions are not really about LAMMPS but about your research and the science you are aiming to do. This makes your questions are topic for a thorough research of the published literature and for a discussion with your adviser or tutor or similar. We can help with “technical” problems with LAMMPS, but to answer your questions in a meaningful way, one would have to be an expert in your specific application of LAMMPS rather than of LAMMPS itself. This is not very likely to find among the subscribers of this forum, let alone the small subset of those that are responding to posts here.
Thank you for taking the time to reply.
Your answers are always very helpful to me.
I apologize that my question did not align with the purpose of the forum.
As Axel said, this is not the place to guide your research. Nor, you want to follow random advice from a forum.
One of the things that took me longer to appreciate as a beginner was how varied is the LAMMPS toolbox. The documentation is excellent, but if you don’t know what to look for, it’s hard to figure out a technical solution --or to even imagine that one may be available.
That said, your script is well organised and there are no obvious flaws that I can think of. The results you get are, as you rightly concluded, a likely consequence of the simulation choices made. In your case, the intermolecular interactions (modelled with a lj/cut pair potential) will affect the tensile strain induced by the interaction with the surface. I too think that the model parameters are likely responsible for the observed mismatch with the experimental evidence. How did you obtain those parameters? Fitting accurate intermolecular interactions could be a line worth pursuing.
You can quickly investigate the effect of the intermolecular interactions on the computed tensile strain by incrementally increasing the strength of LJ epsilon parameter.
In LAMMPS, this is easily achieved by creating a loop and using fix adapt, as in this snippet:
# Increase to a factor 10 linearly
variable i loop 3 20
label increase_strenght
print "*** Increasing strength by a factor $(i/2.) ***"
dump TRJ all 1000 simulation${i}.dump atom # or whatever you need.
variable eps13 equal 0.01462*$i/2.
variable eps14 equal 0.00661*$i/2.
variable eps23 equal 0.0059625*$i/2.
variable eps24 equal 0.0026956*$i/2.
fix STICK all adapt 0 pair lj/cut epsilon 1 3 v_eps13 &
pair lj/cut epsilon 1 4 v_eps14 &
pair lj/cut epsilon 2 3 v_eps23 &
pair lj/cut epsilon 2 4 v_eps24
run ${relax_run}
undump TRJ
unfix STICK
next i
jump SELF increase_strenght
variable i delete
You may want to add more commands in the loop, such as saving the output to a different log file with the log command or running all simulations from the same starting configuration. Also, read the documentation of the commands you are not familiar with. Good luck!
I agree with the other replies that your first post was very thoughtful and comprehensive. I am only commenting on this section:
If you are posing this as a kinetic hypothesis (that is, relaxation during preparation dynamically “heals” the lattice mismatch in a physically unrealistic way), then you could investigate this by “freezing” the MoS2 during equilibration. This is as simple as applying the integrator (fix nvt) only to the substrate instead of to all atoms.
I am not saying this is a scientifically plausible (or not plausible) hypothesis, but from the technical standpoint of using LAMMPS that’s how you can investigate it.
Be careful. I am just suggesting a way to alter the intermolecular interactions using LAMMPS syntax. The scientific problem here is to accurately model those interactions, and it is your job to provide a justification and rationale for the choices made.
Given your excellent starting point, I am sure you will have no trouble setting up numerical experiments to test your working hypotheses, as @srtee suggested.