Please reply to the list.
And, yes.
Please reply to the list.
And, yes.
Ray
I’m sorry for replying on your id.
Still, I am not getting why it’s not (-3 1 0). I have attached a file for calculating miller indices for vectors after rotation.
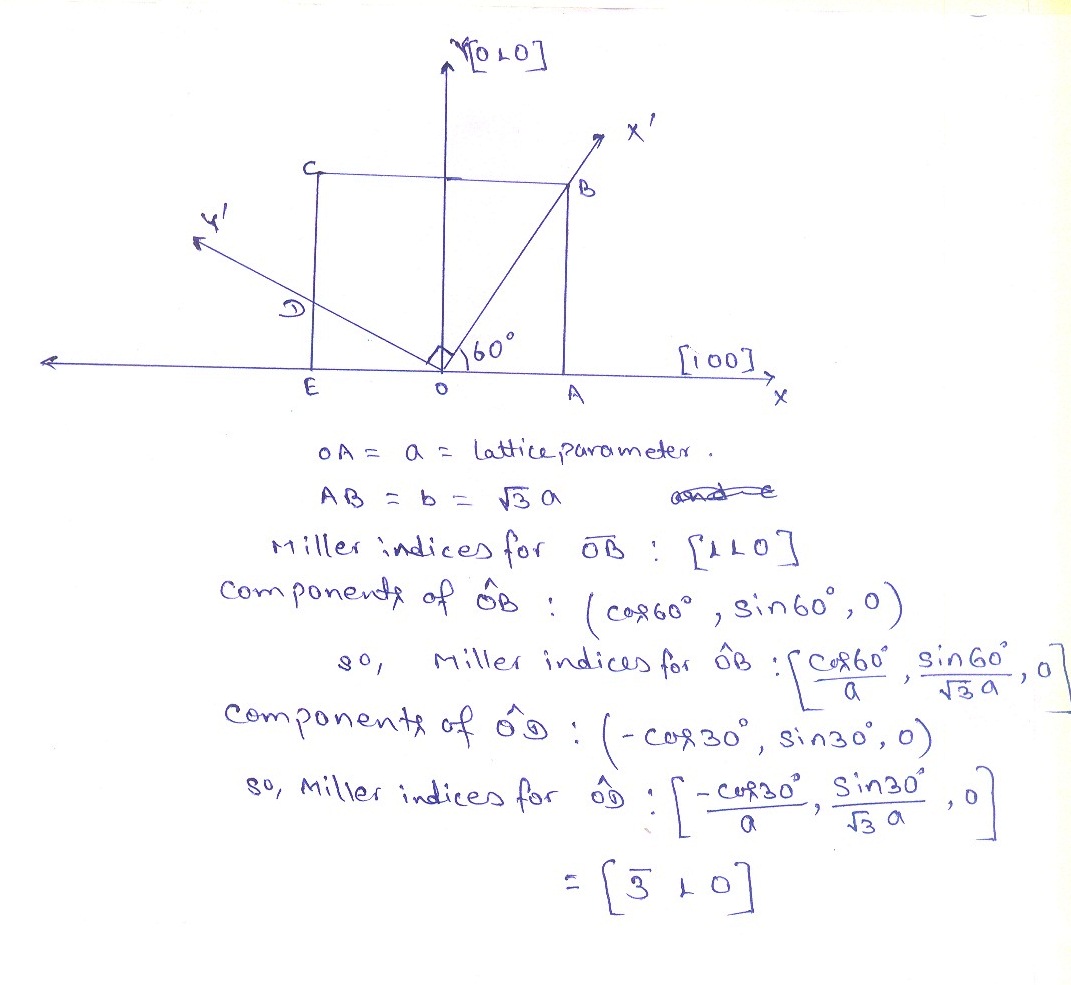
Sorry to say to you that your calculation is still incorrect. My question to you is: what is the angle between [100] and [110]? 60 degrees? or something else? Figure this out.
Ray
Yes, I know if it’s cubic unit cell, then angle will be 45°. But for this case, I already said that unit cell is orthorhombic ( a, b and c are distinct and intersect at 90° angles), so it will be 60°.
No, definitely not. You are mixing “orientation” with “magnitude”. The ultimate check is if the dot product is zero, then they are orthogonal, if not, then not orthogonal.
I have already explained this quite a few times, and since this is really not a LAMMPS Q but a crystallography Q, I will stop here and let you figure this out on your own.
Ray