Here two questions confused me, I would be very grateful if you could give me some guidance.
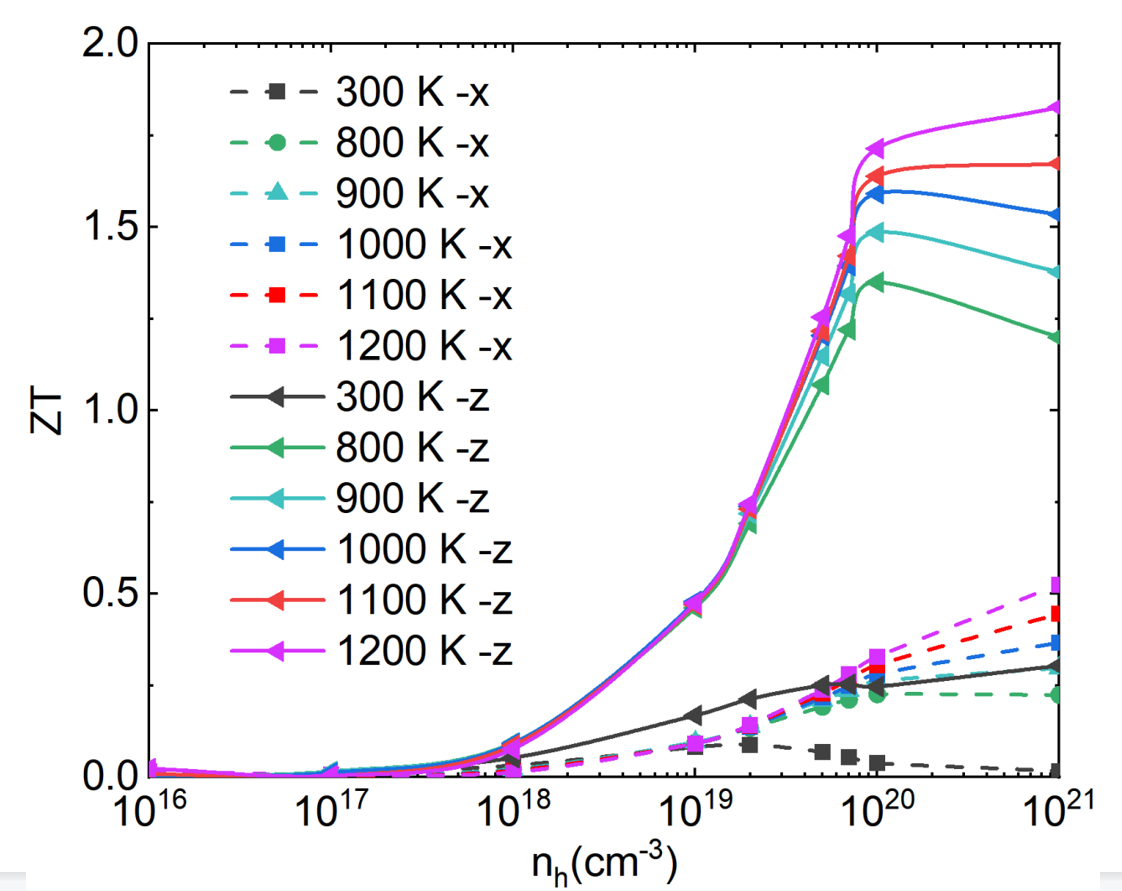
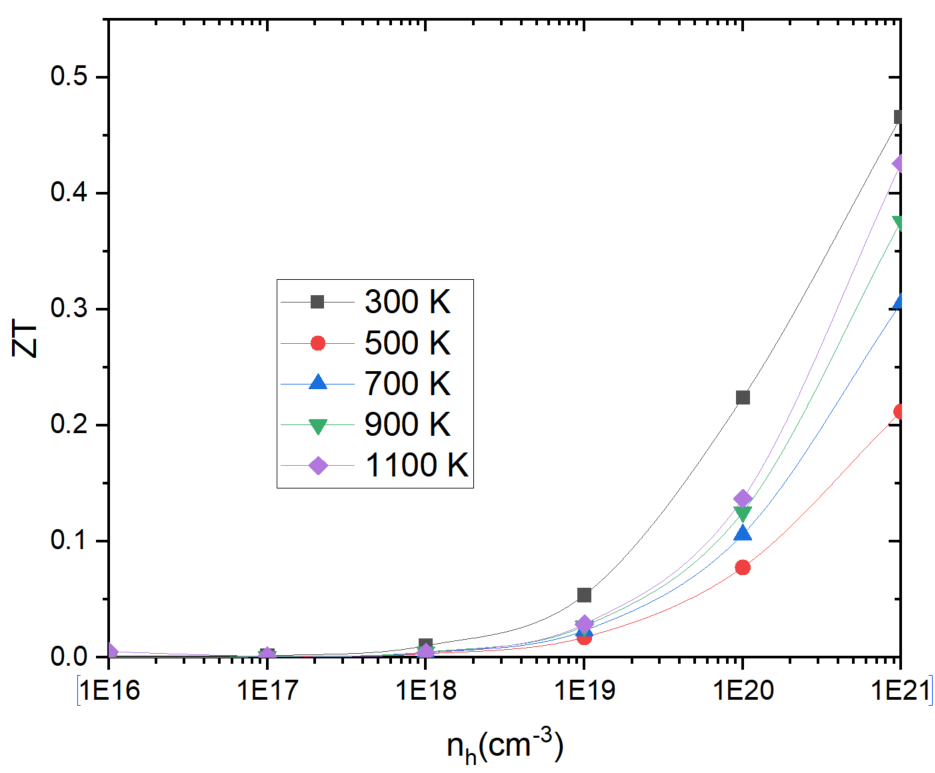
The first question is when I calculated a kind of high temperature thermoelectric material, with the temperature increasing, the ZT value of hole doping does’t appear the maximum value with the increase of concentration, as shown in fig.1, the electron doping is very normal. However, in another simiconductor material,the ZT value of hole doping was very strange too, as shown in fig.2. I can’t understood why we got this result.
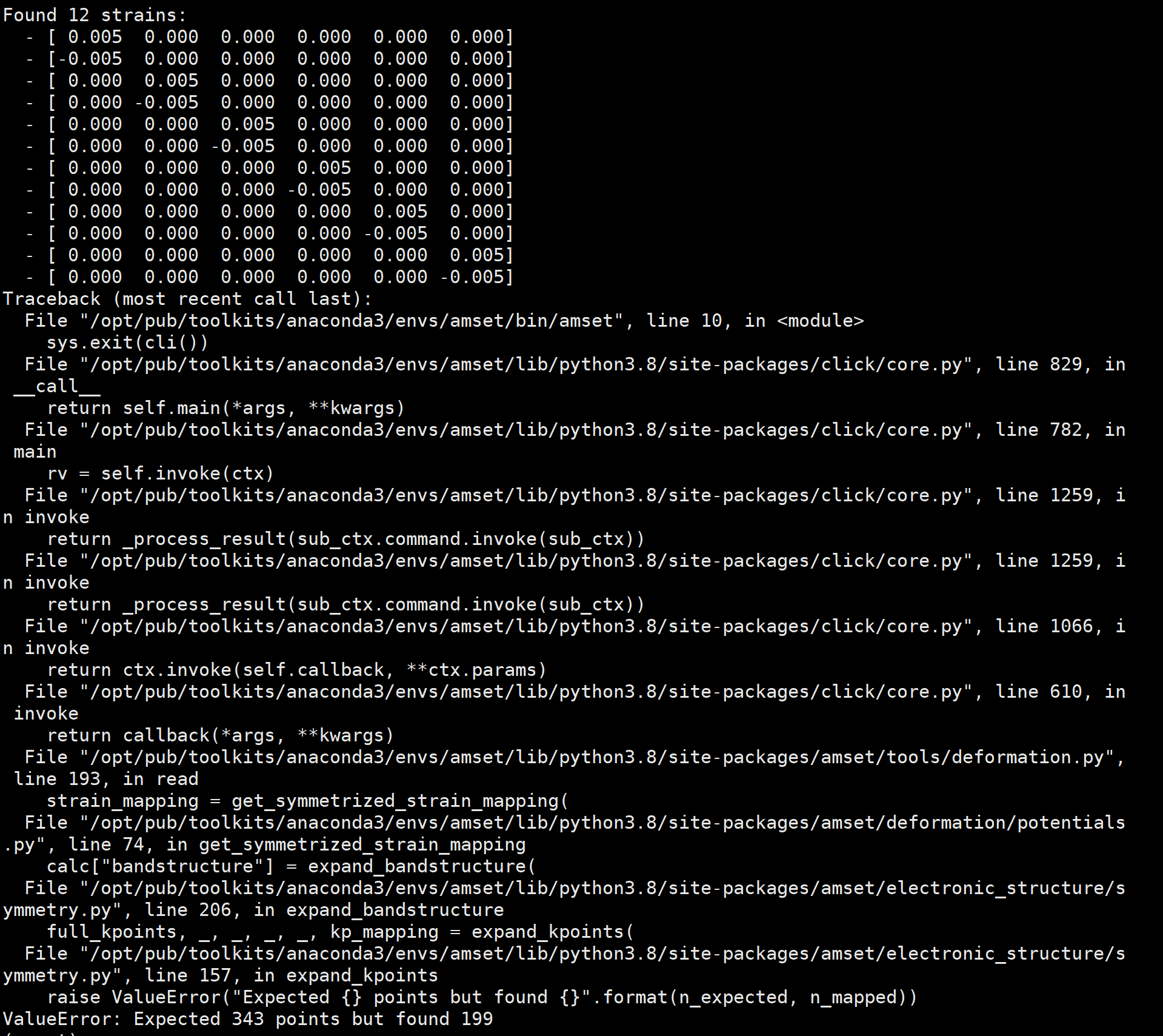
The second question is when I calculating a kind of low-symmetry semiconductor, I run ‘amset deform create’ command, produce twelve different POSCAR structure(code reminding me considering symmetric -N, without consideing this reminding, only generate six different structure), a 777 KPOINTS was used to VASP calculating, when I generated deformation.h5, it reporting an error called ‘ValueError: Expected 343 points but found 199’, as shown in fig.3, I checked the Materials Science forum and github can’t got an good solution.
Best wishes to you, I would appreciate it if you could help me.